Sturge-weber Syndrome with Bilateral Fronto-parieto-temporal Lobe Atrophy and Bilateral Leptomeningeal Angiomatosis: A Rare Case
By Ashok Kumar, Imtiaz Ali Panhwar, Bushra Rehan, Kanchan ShadaniAffiliations
doi: 10.29271/jcpsp.2022.08.S136ABSTRACT
Sturge-weber Syndrome (SWS) is a phacomatosis characterised by Port-Wine stains, leptomeningeal angiomatosis, and glaucoma. Leptomeningeal angiomatosis is seen in 10% to 20% of the cases with the facial nevus, usually on the ipsilateral side. Parietal and occipital regions are the most commonly involved areas; however, it can involve any area of the brain. Bilateral involvement is detected in 15% of the patients. Bilateral Port-Wine stains have been shown to be associated with an increased risk of developing epilepsy in an early age. Here, we present a case of a 3-month child with bilateral Port-Wine stains who presented with generalised fits. On the basis of Port-Wine stains, SWS was suspected, and MRI was done which showed cerebral atrophy in bilateral fronto-parieto-temporal regions and bilateral leptomeningeal angiomatosis, which are rare findings according to the literature.
Key Words: Sturge-weber syndrome, Phacomatosis, Port-wine stain, Fits.
INTRODUCTION
Sturge-weber Syndrome (SWS) is a phacomatosis (a group of neurocutaneous disorders involving structures that arise from the embryonic ectoderm) characterised by a Port-Wine stain mostly involving that part of the face which is innervated by 1st and 2nd division of trigeminal nerve and malformations of veins of the brain and eyes.1-4
Although SWS is sporadic in nature, but some familial cases have also been reported. In recent studies, it is discovered that SWS is associated with a mutation in the gene called guanine nucleotide binding protein G (q) subunit alpha, human (GNAQ).2,5 Basic cause of the disease is unknown till date.1
Clinical features include Port-Wine stain, epilepsy, glaucoma, visual impairment, migraine, hemiparesis, and cognitive impairments.4
Imaging findings include cortical hemiatrophy, gyral calcifications usually after 2 years of age, choroid plexus enlargement, cranial diploe prominence, and pial angiomas.
Bilateral involvement is seen in 15% of the patients,1,3 which results in increased susceptibility of having seizures that begin earlier than in patients having unilateral disease.6 We present a case of a 3-month child with bilateral Port-Wine stains who presented with generalised fits. On the basis of Port-Wine stains, SWS was suspected, and MRI was done which showed cerebral atrophy in bilateral fronto-parieto-temporal regions and bilateral leptomeningeal angiomatosis, which are rare findings according to the literature.1
CASE REPORT
A 3-month male child presented with complaints of generalised fits and apneic spells. He has the previous history of 3 episodes of fits in the past which subsided on their own.
On examination, the patient had extensive Port-Wine stains bilaterally (Figure 1). He was irritable but was tolerating the mother’s feed. He was vitally stable with no history of fever. Routine laboratory tests were in the normal range. Electroencephalograph (EEG) was normal.
The MRI revealed atrophy of bilateral fronto-parietal and temporal lobes, more on the right side (Figure 2). There was a significant gyral enhancement of the right frontal, and bilateral parieto-temporal regions and mild gyral enhancement of the left occipital region (Figure 3). There was the inhomogeneous enhancement of tiny vessels in extra-axial CSF space along bilateral fronto-parietal and temporal regions, more marked on the right side suggestive of leptomeningeal angiomas.
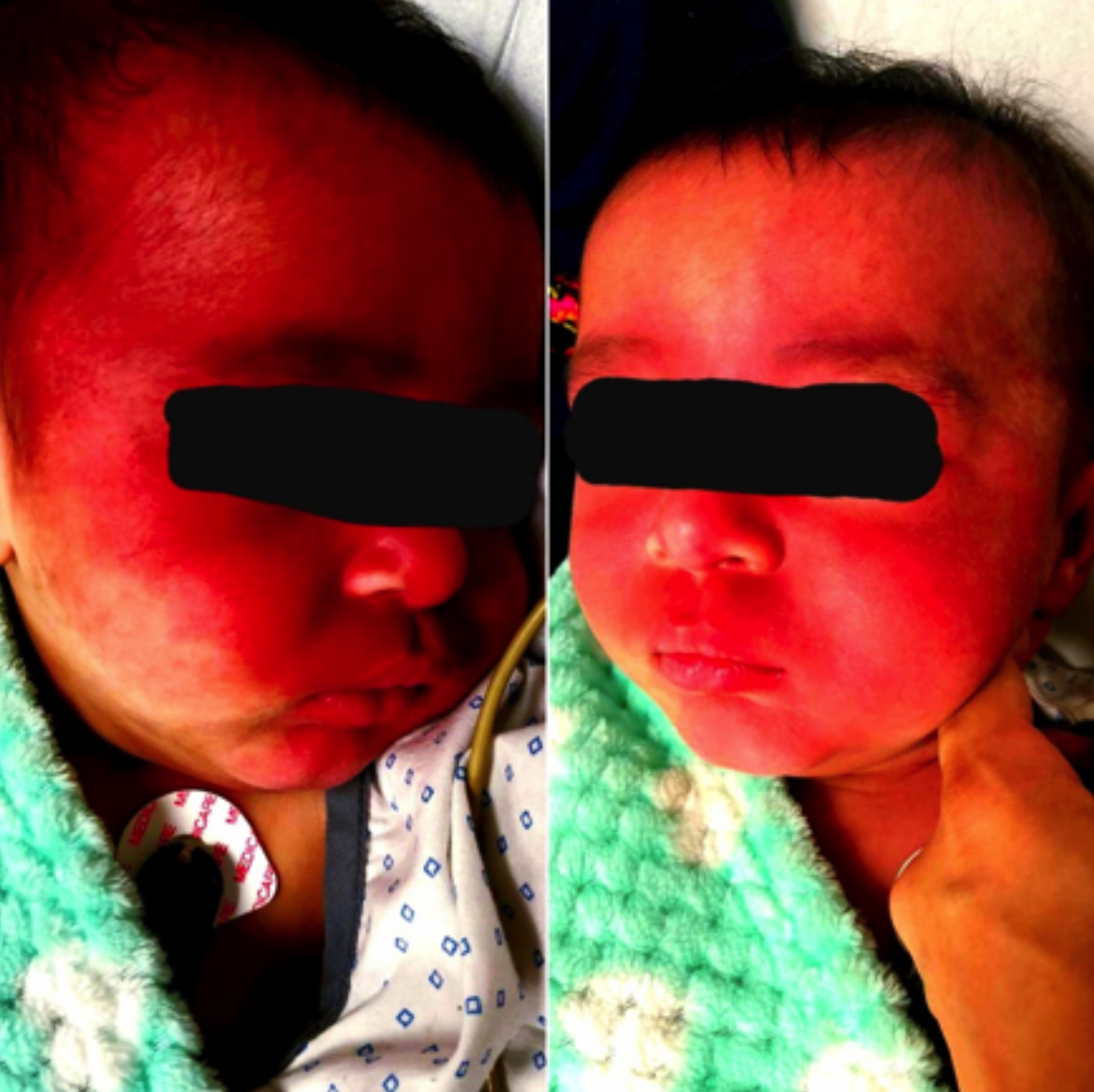 Figure 1: Extensive port-wine stains bilaterally.
Figure 1: Extensive port-wine stains bilaterally.
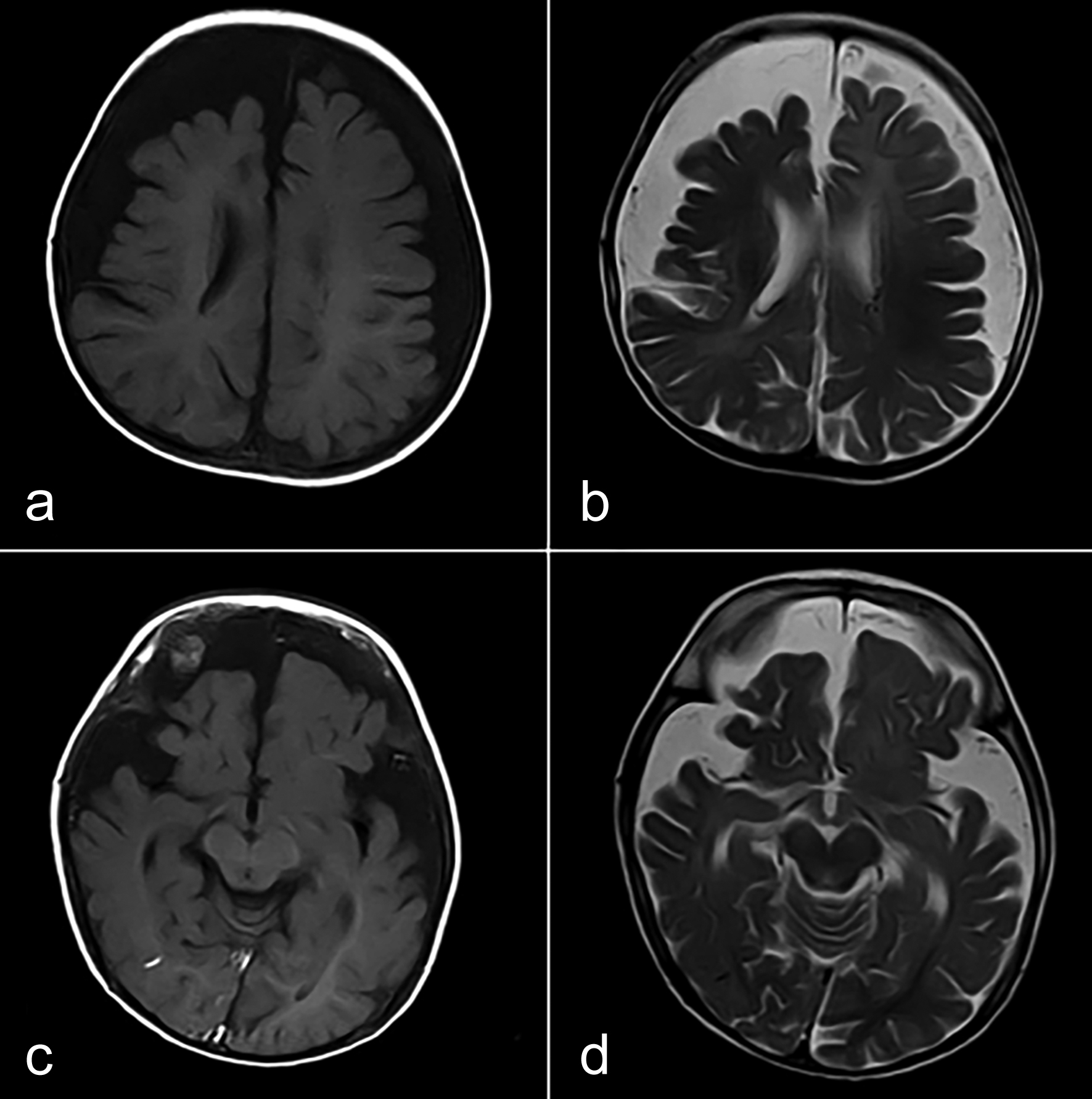 Figure 2: (a,b) Atrophy of bilateral fronto-parietal lobes, more on the right side. (c,d) Atrophy of bilateral temporal lobes and normal occipital lobes.
Figure 2: (a,b) Atrophy of bilateral fronto-parietal lobes, more on the right side. (c,d) Atrophy of bilateral temporal lobes and normal occipital lobes.
On the basis of these clinical and radiological findings, the diagnosis of SWS was made involving bilateral cerebral hemispheres with bilateral leptomeningeal angiomas.
DISCUSSION
SWS was initially described by an ophthalmologist Schirmer in 1860. William Allen Sturge described the cutaneous, ocular, and neural features of the disease in 1879. In 1922, Parkes Weber documented the radiological changes observed in these patients.1
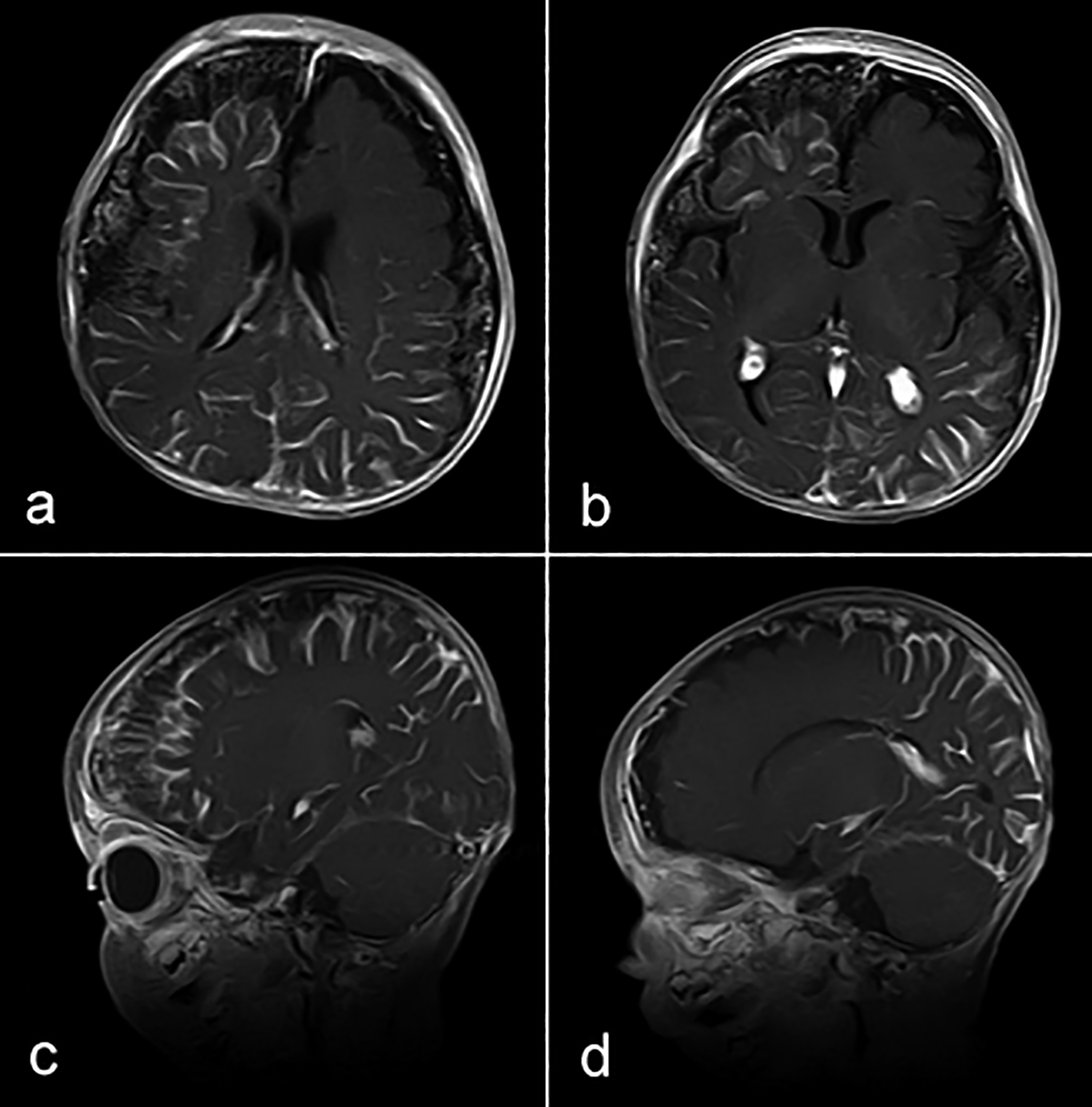 Figure 3. Gyral enhancement of right frontal, bilateral parieto-temporal regions (a,b) and mild gyral enhancement of left occipital region (b,d). There are leptomeningeal angiomas along bilateral fronto-parietal and temporal regions (a,b,c,d ).
Figure 3. Gyral enhancement of right frontal, bilateral parieto-temporal regions (a,b) and mild gyral enhancement of left occipital region (b,d). There are leptomeningeal angiomas along bilateral fronto-parietal and temporal regions (a,b,c,d ).
Although the exact cause is unknown till date but the basic abnormality in this entity seems to be the lack of thrombosis of superficial veins of the cortex on the diseased side. This results in redirection of blood with the formation of pial angiomas and engorgement of the deep veins. The diversion of venous blood does not compensate fully, so there is stasis of blood resulting in chronic hypoxia which ultimately result in loss of brain tissue and dystrophic gyral calcifications. These are the factors that are responsible for radiological features in SWS such as cerebral atrophy, gyral calcification, and leptomeningeal enhancement.7
Most commonly patients present with a seizure in infancy. Bilateral hemispheric involvement results in increased susceptibility to having seizures that begin earlier than in patients having unilateral disease.5,7 Stroke-like episodes are more common in young children which are likely due to frequent falls resulting in brain damage. Headaches, migraine, attention deficit and learning disabilities may also occur in patients of SWS.8
Ocular manifestations include glaucoma, buphthalmos, and retinal and choroidal detachments. Homonymous hemianopia can also occur which is due to angiomatosis in both occipital regions affecting the adjacent brain. Other ocular manifestations include choroidal hemangiomas and vascular malformations in the episcleral lamina or conjunctiva.1
MRI is currently the imaging investigation of choice with pial enhancement on post-contrast study considered the most reliable finding in assessing the extent of disease.2,5 Unenhanced CT should be used only if MRI is normal, to see the presence of gyral calcifications. Imaging findings include cortical hemiatrophy, gyral calcifications usually after 2 years of age, choroid plexus enlargement, cranial diploe prominence, and pial angiomas. The white matter, which is below the damaged cortex, appears hyperintense on T2-weighted (T2W) images which is due to the ischemic and gliotic changes.
In conclusion, this case showed bilateral imaging findings associated with SWS, which are rare and are only seen in 15% of cases and result in increased susceptibility to having seizures that begin earlier than in patients having unilateral disease. Therefore, prompt and correct diagnosis is critical in the management of these patients.
PATIENT’S CONSENT:
Informed consent was taken from the patient’s parents to publish the data with disclosing the name.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
AK: Collected patient data and wrote the case report.
IAP, KS: Reviewed the current literature and previously reported cases.
BR: Supervised and reviewed the final text of the case.
REFERENCES
- Pérez AM, Rojas MR, Martín VP, Carral JD, Sáez IC, Rodríguez AD, et al. Analysis of sturge weber syndrome: A retrospective study of multiple associated variables. Neurología (English Edition). 2017; 32(6):363-70. 10. doi: 1016/j.nrleng.2015.12.006.
- Bar C, Pedespan JM, Boccara O, Garcelon N, Levy R, Grévent D, et al. Early magnetic resonance imaging to detect presymptomatic leptomeningeal angioma in children with suspected Sturge–weber syndrome. Dev Med Child Neurol 2020; 62(2):227-33. doi: 10.1111/ dmcn.14253.
- Saeed MA, Hilal K, Chand P. Bilateral intracranial calcifications with bilateral facial cutaneous naevus: Sturge weber syndrome. Case Rep 2017; 2017. doi: 10.1136/bcr-2017-219985.
- Kaseka ML, Bitton JY, Décarie JC, Major P. Predictive factors for epilepsy in pediatric patients with sturge weber syndrome. Pediatric Neurol 2016; 64(2):52-8. doi: 10.1016/j.pediatrneurol.2016.08.009.
- Adams ME, Aylett SE, Squier W, Chong W. A spectrum of unusual neuroimaging findings in patients with suspected Sturge-weber syndrome. Am J Neuroradiol 2009; 30(2):276-81. doi: 10.3174/ajnr.A1350.
- Bebin EM, Gomez MR. Prognosis in sturge-weber disease: Comparison of unihemispheric and bihemispheric involvement. J Child Neurol 1988; 3(3):181-4. doi: 10.1177/088307388800300306.
- Mandelstam S, Andronikou S. MRI evaluation of venous abnormalities in children with Sturge-weber syndrome. J Pediatric Neurol 2004; 2(01):029-32. doi: 10.1055/s- 0035-1557187.
- Comi AM. Presentation, diagnosis, pathophysiology and treatment of the neurologic features of sturge-weber syndrome. Neurologist 2011; 17(4):179. doi: 10.1097/ NRL.0b013e318220c5b6.