Novel Mutation of Hydroxymethylbilane Synthase in a Case of Acute Intermittent Porphyria Presenting with Posterior Reversible Encephalopathy Syndrome
By A Li Yang, Li Min Ma, Hong Ju Zhang, Jie Wen ZhangAffiliations
doi: 10.29271/jcpsp.2022.Supp.S102ABSTRACT
Acute intermittent porphyria (AIP) is an autosomal, dominant, hereditary metabolic disease caused by an inherited deficiency of hydroxymethylbilane synthase (HMBS), a crucial enzyme in the heme biosynthetic pathway. It can affect the central, peripheral, and autonomic nervous systems. We report a 23-year Chinese woman who presented with severe abdominal pain, convulsions, constipation, tachycardia, quadriparesis, and hyponatremia, accompanied by posterior reversible encephalopathy syndrome (PRES). The clinical diagnosis of AIP was made after positive urine Watson–Schwartz test for porphobilinogen (PBG). Genetic testing is important for AIP patients in confirming the diagnosis. We identified a new insertion mutation in intron 14 [c.1005dupC (p.I336Hfs*23)] of the HMBS in her genomic DNA. Timely and accurate treatment of AIP may improve disease prognosis.
Key Words: Acute intermittent porphyria, Mutation, Posterior reversible encephalopathy syndrome.
INTRODUCTION
Acute intermittent porphyria (AIP), as the most commonly seen acute porphyria, is an autosomal, dominant, hereditary metabolic disorder resulting from the mutation of hydroxymethylbilane synthase (HMBS) gene, a crucial enzyme in the heme biosynthetic pathway.1 The diagnosis of AIP is not easy due to highly variable and nonspecific clinical features. Posterior reversible encephalopathy syndrome (PRES) is associated with bilateral posterior white and/or grey matter lesions that present with headaches, visual disturbances, seizures, and mental disturbances.2 Here we describe a case of AIP with a novel HMBS gene mutation that presented with PRES.
CASE REPORT
A 23-year Chinese female was admitted to our centre for intermittent constipation accompanied by abdominal pain and convulsions that lasted for more than four months. Initially, the diagnosis was intestinal obstruction. The generalised tonic-clonic seizures were considered to be an adverse effect of the Primperan used in the first round of treatment.
However, the seizures occurred repeatedly as the disease progressed. In addition, abnormal signals were found using brain magnetic resonance imaging (MRI). The patient also described recurrent, agonizing, paroxysmal abdominal, and femoral pain after admission. Particular medical information and family history were denied. Her clinical examination was indistinctive, except for a mild, symmetrically decreased myodynamia and tendon reflexes in the limbs. The patient had slight tachycardia (110/min) and critical blood pressure (BP) (139/84 mmHg). Her haemoglobin (Hb) was 100 g/L (Normal: 110-150 g/L) and serum sodium was 115 mmol/L (Normal: 137-147 mmol/L). Thyroid function tests suggested low T3 syndrome. Several air-fluid levels were found in her abdominal x-ray images. In the brain MRI in bilateral occipital, parietal, and frontal lobes, there were multifocal lesions which were hypointense on T1-weighted images and hyperintense on T2-weighted and fluid-attenuated inversion recovery (Flair) images. Isointense to hypointense states in diffusion-weighted images (DWI) and a hyperintense state in apparent diffusion coefficient (ADC) maps suggested vasogenic oedema rather than cytotoxic oedema in PRES. Her abdominal pain was recurrent and severe without clear aetiology; her urine was with mysterious colour that changed to dark and red upon exposure to sunlight. These findings led us to consider AIP. The clinical diagnosis of AIP was confirmed after the patient’s urine tested positive for porphobilinogen (PBG) using the Watson–Schwartz test. Glucose infusion and symptomatic treatment were given to the patient, as hematin was not available in our district. With these treatments, the patient’s condition improved gradually. Repeated brain MRI demonstrated that the bilateral lesions reduced six days later, which is compatible with PRES.
The patient and her parents signed a written consent form and accepted genetic testing. A novel HMBS gene (NM_000190.4) insertion mutation, c.1005dupC (p.I336Hfs*23), was detected in the patient by Sanger sequencing. Structural changes in this gene likely affect the activity and stability of the predicted protein. In addition, according to the Genetic Variant Interpretation Tool, the interpretation of the detected mutation was “pathogenic,” which led us to the speculation that the novel mutation is likely related to the patient’s AIP attack. The patient recovered completely and returned to work six months later, and no recurrence has been reported so far.
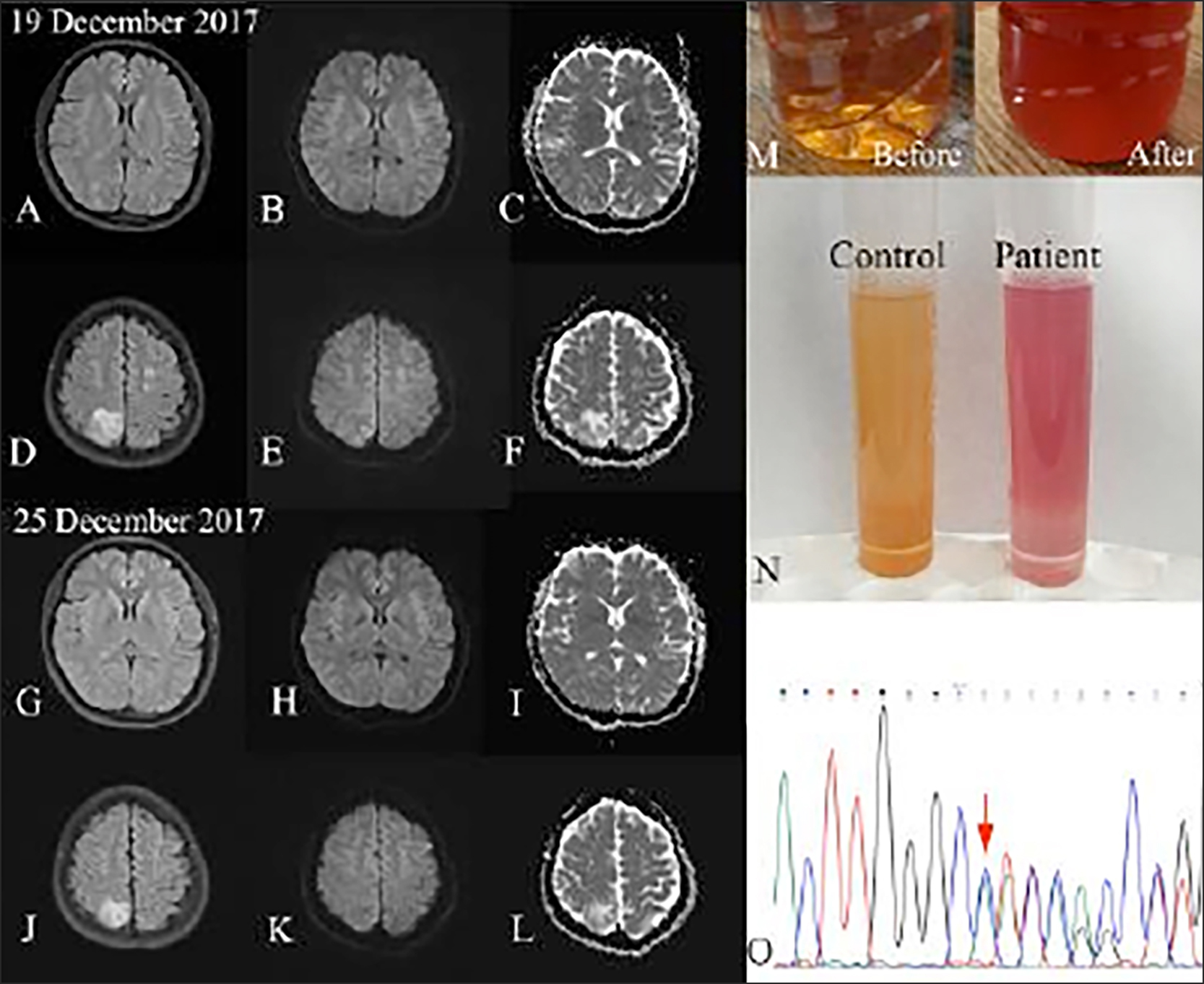 Figure 1: MRI shows lesions on Flair images(A,D,G,J), DWI images (B,E,H,K) and ADC maps (C,F,I,L). Sunlight test (M) and Watson-Schwartz test (N) suggested raised urinary PBG. A novel insertion mutation of HMBS gene was identified in the patient (O).
Figure 1: MRI shows lesions on Flair images(A,D,G,J), DWI images (B,E,H,K) and ADC maps (C,F,I,L). Sunlight test (M) and Watson-Schwartz test (N) suggested raised urinary PBG. A novel insertion mutation of HMBS gene was identified in the patient (O).
DISCUSSION
Precipitation of AIP is related to many diverse factors, such as drugs, steroid hormones, diet, menstruation, infection, surgery, and stress. Symptoms caused by excessive production of porphyrin precursors of AIP are intermittent, and sometimes life-threatening.3 Clinical manifestations in the central nervous system (CNS), including epileptic seizures, disturbance of consciousness, and hyponatremia, are caused by inappropriate antidiuretic hormone syndrome. Peripheral and autonomic neuropathy are more frequent and include weakness, sensory changes, abdominal pain, tachycardia, hypertension, nausea, and vomiting. General weakness can sometimes develop rapidly into quadriparesis and acute respiratory paralysis, especially when patients have been misdiagnosed and treated with barbiturates or hydantoins. Our patient’s symptoms included constipation, abdominal and femoral pain, tachycardia, quadriparesis, seizures, and hyponatremia.
PRES has been reported in very few patients with AIP. Here, we described a new AIP case with PRES. In this case, abnormal signals in the cerebral cortex were found on the MRI. As observed in this patient, the lesions are usually totally or partially reversible and there are symmetric cortical and subcortical involvements of the occipital and parietal lobes, without or with mild enhancement. Although PRES often presents as vasogenic oedema caused by hypertension, there are still more causes that involve factors like eclampsia, autoimmune disorders, uremia, malignancies, and peripartum states, to name a few.
The notably increased urinary excretion of accumulated metabolites, including PBG and aminolevulinic acid (ALA), is of great importance for definitive diagnosis. The Watson–Schwartz test4 can provide clues when further quantitative biochemical investigations are unavailable. Molecular genetic testing offers an accurate diagnosis and typing for symptomatic patients, which can then be applied to the identification of AIP among relatives of the proband. So far, more than 400 AIP-related mutations in the HMBS gene have been reported. The authors of this study detected a novel small insertion mutation located at exon 14 in the HMBS gene. Regrettably, the patient’s family refused genetic testing. They did receive information about the risk factors that increase the likelihood of an attack, such as certain drugs or other environmental factors. Pertinent symptomatic treatment is important for the management of AIP and the efficacy of heme therapy has been validated as an efficacious treatment.5 Considering potential deficiency or adverse effects of preceding methods, additional approaches to therapy, such as liver transplantation and small inhibitory RNAs (siRNAs), are being explored.
In summary, AIP should be taken into consideration in the differential diagnosis of PRES and genetic testing is important for suspected AIP patients in confirming the diagnosis. Timely and accurate treatment of AIP may also improve disease prognosis.
PATIENT’S CONTENT:
Informed consent to publish this case was obtained.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
ALY, LMM: Literature search, data acquisition, data analysis, manuscript preparation, and manuscript editing.
HJJ, JWZ: Design of intellectual content, literature search, and manuscript review.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute hepatic porphyrias: Review and recent progress. Hepatol Commun 2019; 3(2):193-206. doi: 10.1002/hep4.1297.
- Brady E, Parikh NS, Navi BB, Gupta A, Schweitzer AD. The imaging spectrum of posterior reversible encephalopathy syndrome: A pictorial review. Clin Imaging 2018; 47:80-9. doi: 10.1016/j.clinimag.2017.08.008.
- Besur S, Schmeltzer P, Bonkovsky HL. Bonkovsky. Acute porphyrias. J Emerg Med 2015; 49(3):305-12. doi: 10.1016/j.jemermed.2015.04.034.
- Schreiber WE, Jamani A, Pudek MR. Screening tests for porphobilinogen are insensitive. The problem and its solution. Am J Clin Pathol 1989; 92(5):644-9. doi: 10.1093/ajcp/92.5.644.
- Anderson KE,Collins S. Open-label study of hemin for acute porphyria: Clinical practice implications. Am J Med 2006; 119(9):801.e19-24. doi: 10.1016/j.amjmed.2006.05.026.