Lafora Body Epilepsy: A Challenging Diagnosis
By Ali Zohair Nomani1, Haris Majid Rajput2, Hanna Nomani3Affiliations
doi: 10.29271/jcpsp.2022.08.S133ABSTRACT
Lafora body disease (LBD) is a progressive myoclonic genetic epilepsy syndrome characterized by the presence of Lafora inclusion bodies within neurons and other cells. It is a complex neurodegenerative disease presenting in adolescence with seizures, myoclonus, and rapid cognitive decline. Diagnosis is often challenging requiring a thorough history including family history, identification of Lafora bodies in apocrine sweat glands of axillary skin, and specific DNA sequencing. There is no cure and management is mainly supportive. We present one of the only few cases from Pakistan of LBD based on characteristic biopsy findings, history of similar ailment in siblings, and EPM2B mutation. This case emphasizes the need for physicians and neurologists to be aware of diagnostic challenges associated with LBD and its characteristic findings.
Key Words: Lafora body, Progressive epilepsy, Myoclonus, Axillary skin biopsy, EPM2B.
INTRODUCTION
Lafora progressive myoclonus epilepsy (PME) is a brain disorder characterised by the recurrent seizures, sometimes, refractory, and a progressively declining cognition.1 The signs and symptoms of the disease usually manifest in late childhood or adolescence.1 Typical age of presentation is between 8 to 19 years and the disease quickly worsens.1,2
PME accounts for about 1% of all the epilepsies.2,3 Lafora body disease (LBD) is one of the common PMEs. It is distinguished by the presence of inclusion bodies called "Lafora bodies" found inside the cytoplasm of a variety of cell types including neurons, cardiac myocytes, hepatocytes, and skin.1,2
We present a case of a young girl with refractory seizures, progressive cognitive impairment and functional disability diagnosed as Lafora body disease. This case highlights the classical skin findings and genetic mutations of LBD in a Pakistani family and emphasizes the need for physicians and neurologists to be aware of diagnostic challenges associated with LBD.
CASE REPORT
A 17-year girl was hospitalised at our centre with 3 years history of the right-sided simple partial seizures and occasional myoclonic seizures. Her two brothers died of secondary to similar ailments at the age of 17 years after being sick for 5 years. Our patient showed behavioral changes culminating in functional disability, irritability, and poor ability to understand and follow simple commands. Initially, she had difficulty walking until she became bed-bound and started experiencing frequent hallucinations. She had previously been prescribed sodium valproate, lamotrigine, and carbamazepine at different time points. Her seizures progressed to generalised tonic-clonic episodes, and their frequency increased until she presented with status epilepticus (SE).
Given the course of her disease, we performed an extensive diagnostic evaluation to ascertain the cause of seizures. Magnetic Resonance Imaging (MRI) showed diffuse brain atrophy but otherwise nonspecific white matter changes (Figure 1). Cerebrospinal fluid analysis and metabolic tests including autoimmune panel were unremarkable. Electroencephalogram showed paroxysmal polyspike waves lateralized to left temporal leads. An extended work-up by doing an axillary skin biopsy revealed Lafora bodies on Periodic acid-Schiff (PAS) stain as shown in Figure 2.
To characterise associated genetic mutation, DNA extracted from a peripheral blood sample was processed using a gentra puregene blood DNA purification kit (Qiagen Inc., Valencia, CA, USA). Our analysis identified the causative mutation for her LBD in the EPM2B gene (homozygous) (Figure 3). Blood samples from parents were analysed revealing both to be heterozygous carriers of the EPM2B gene mutation.
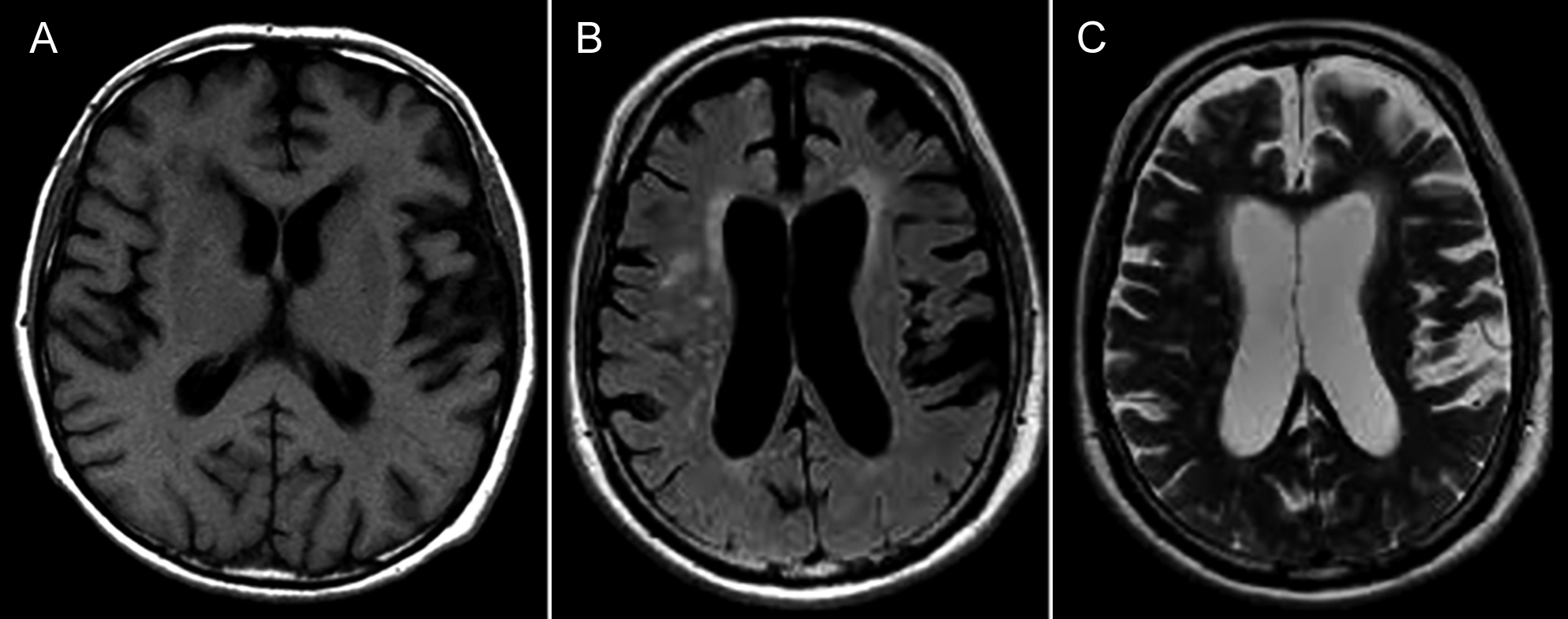 Figure 1: MRI Brain showing non-specific white matter hypointensities on T1 (A), and hyperintense signals on FLAIR (B) and T2 (C) with cerebral atrophy.
Figure 1: MRI Brain showing non-specific white matter hypointensities on T1 (A), and hyperintense signals on FLAIR (B) and T2 (C) with cerebral atrophy.
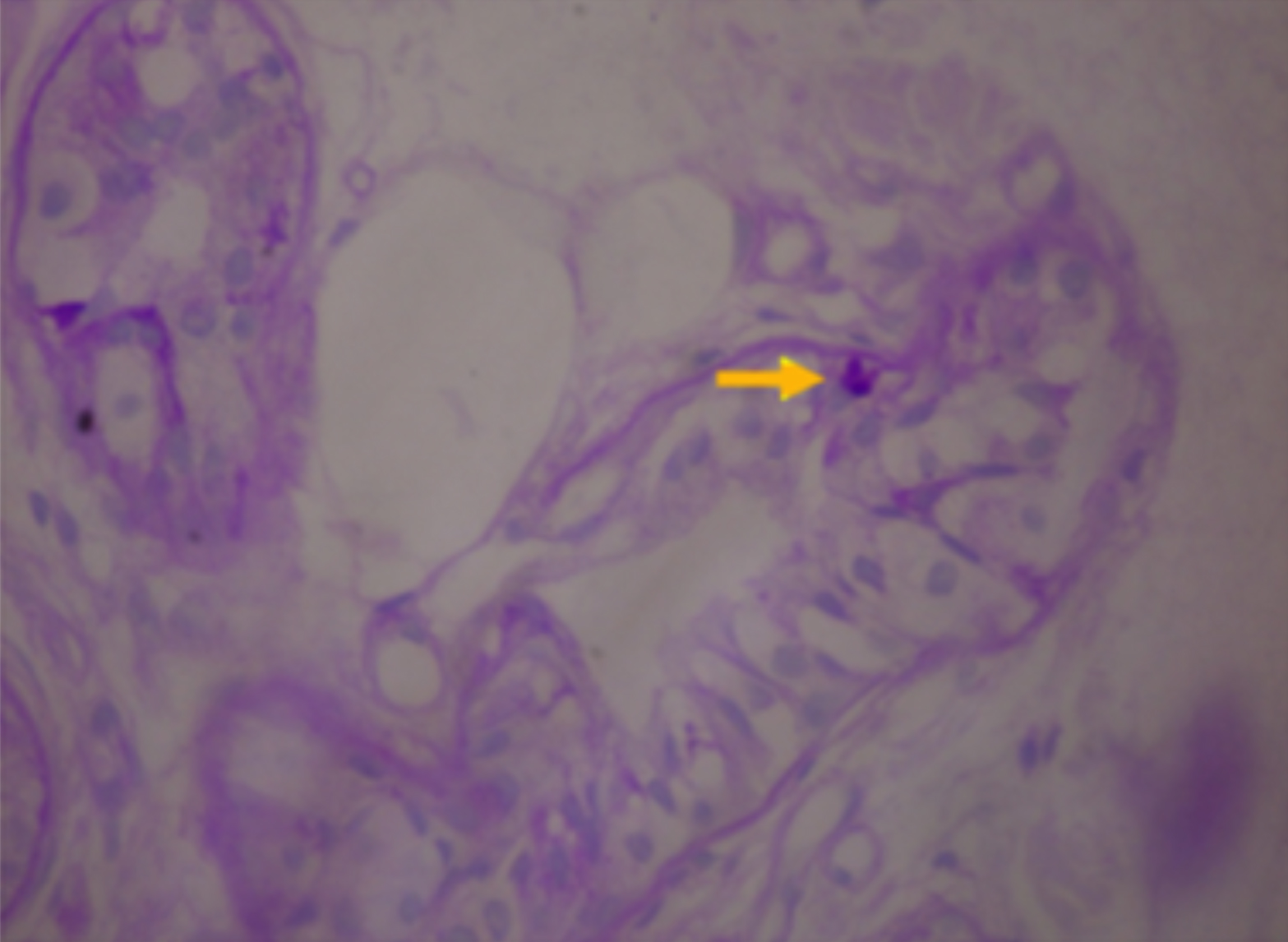 Figure 2: Axillary skin biopsy showing PAS-positive Lafora body (arrowhead) in apocrine glands.
Figure 2: Axillary skin biopsy showing PAS-positive Lafora body (arrowhead) in apocrine glands.
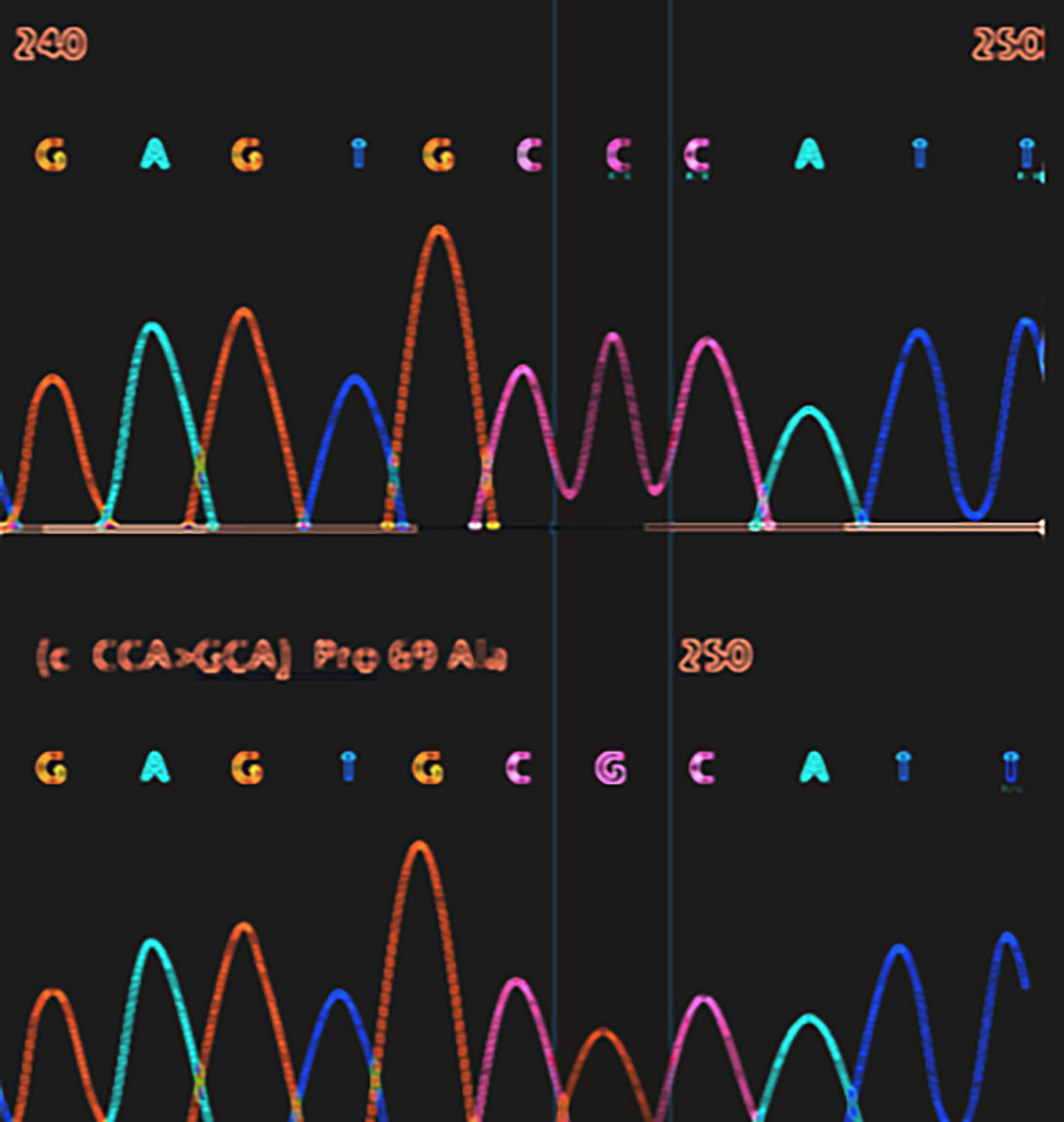 Figure 3: Genetic analysis showed homozygous missense mutation (c205C>G; Pro69Ala) in the EPM2B (NHLRC1) gene as depicted.
Figure 3: Genetic analysis showed homozygous missense mutation (c205C>G; Pro69Ala) in the EPM2B (NHLRC1) gene as depicted.
DISCUSSION
LBD is a complex metabolic neuro-degenerative disorder.1-3 Clinical findings typically begin by end of the first decade or at the start of the second.1,3,4 Majority of patients show reduced life expectancy. With a rapidly declining functional ability, death ensues due to the complications of LBD within ten years of symptoms on average.1,3,4
The exact prevalence of Lafora PME is undetermined.3,4 The disease is most prevalent in the Mediterranean region, parts of Central and South Asia, North Africa, and the Middle East.3,4 Prominent clinical findings include intractable seizures, myoclonus, ataxia, episodes of drop attacks, and most profoundly, a quickly developing and progressive severe cognitive impairment from the time of onset of seizures.2,3 Behavioral sequelae include depression, episodes of confusion, and speech problems as early manifestations.3,4 Subsequently, profound dementia ensues impairing judgment, thinking, and decision making.3 This results in an inability to perform activities of daily living (ADLs) early or in the mid-twenties and eventually leaves patients dependent on full-time care.1-4 Eventually, seizures become refractory to polypharmacy and frequently result in SE.3-5 While the diagnosis of SE is clinical, the preceding course of events leading to SE is of vital significance, as it can lead the physician/ neurologist towards the correct underlying diagnosis.2,4,5 Although the prognosis is grave, refractory seizures specifically myoclonus need to be treated by certain anti-epileptics while avoiding others.4,5
The definite diagnostic tool is a demonstration of Lafora bodies within the apocrine sweat glands retrieved on axillary skin biopsy.4 Carbohydrate enriched Lafora bodies are stained by PAS representing polyglucosans, that precipitate inside cells of various organs including the brain.4,6 Mutations in EPM2A gene or NHLRC1 gene encoding laforin and malin, respectively, account for 80-90% of LBD.2,4-6 Identification of gene mutations has prognostic implications as it can help guide family screening and anticipate potential development of LBD.5,6
In conclusion, refractory myoclonus and rapid cognitive decline should caution physicians to think of unusual and rare diagnoses of PME. Traditional axillary skin biopsy for Lafora bodies is a useful, easy, non-invasive, and specific diagnostic tool that can be supplemented by the identification of specific genetic mutations to aid family screening. This can have clinical implications regarding the judicious and correct use of antiepileptic drugs and explaining prognosis.
PATIENT’S CONSENT:
Written informed consent was obtained from the patient to publish the data concerning this case.
COMPETING INTEREST:
The authors declared no competing interests.
AUTHORS’ CONTRIBUTION:
AZN, HMR, HN: Contributed to the concept, draft the manuscript, and reviewed it critically.
AZN, HMR: Did data collection.
AZN, HN: Prepared the figures.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Al Mufargi Y, Qureshi A, Al Asmi A. Lafora Disease: Report of a Rare Entity. Cureus 2020; 12(1):e6793. doi: 10.7759/ cureus.6793.
- Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease - from pathogenesis to treatment strategies. Nat Rev Neurol 2018; 14(10):606-17. doi: 10.1038/s41582-018-0057-0.
- Desdentado L, Espert R, Sanz P, Tirapu-Ustarroz J. Lafora disease: A review of the literature. Rev Neurol 2019; 68(2):66-74. PMID: 30638256 PMCID: PMC6531605.
- Rhouda H, Malihy A, Zouiri G, Kriouile Y. Pathologic confirmation of lafora disease. Pediatr Neurol 2020; 108: 128. doi: 10.1016/j.pediatrneurol.2020.02.004.
- Martin S, Strzelczyk A, Lindlar S, Krause K, Reif PS, Menzler K, et al. Drug-resistant juvenile myoclonic epilepsy: Misdiagnosis of progressive myoclonus epilepsy. Front Neurol 2019; 10:946. doi.org/10.3389/fneur. 2019.00946.
- Verhalen B, Arnold S, Minassian BA. Lafora disease: A review of molecular mechanisms and pathology. Neuropediatrics 2018; 49(6):357-62. doi: 10.1055/ s-0038-1675238.