Kleefstra Syndrome
By Hilal Aydin1, Ibrahim Hakan Bucak2, Haydar Bagis3Affiliations
doi: 10.29271/jcpsp.2022.04.76ABSTRACT
Kleefstra syndrome (KS), previously referred to as 9q subtelomeric deletion syndrome (9qSTDS), is characterised by moderate to severe developmental delay/mental retardation, childhood hypotonia, and brachy-microcephaly (main clinical phenotype), midface hypoplasia, prognathism, lip and eyebrow shape anomalies. The true prevalence of KS is unknown, but it is estimated that it occurs with a frequency of 1/200.000 in cases with mental retardation. On literature search, approximately 110 patients have been reported so far. Genetic analysis should be planned and interdisciplinary monitoring should be provided in cases suspected to have KS.
Key Words: Child, Genetic disorder, Kleefstra Syndrome, Dysmorphism.
INTRODUCTION
Kleefstra syndrome (KS) is a disorder characterised by mental retardation, childhood hypotonia and peculiar facial dysmorphism. It occurs as a result of deletion on chromosome 9q34.3.1,2 The true prevalence of KS is unknown; however, it is estimated to occur with a frequency of 1/200.000 in cases with mental retardation.3 Typical phenotypic findings highly associated with KS include heart defects, seizures, obesity, eye abnormalities, behavioural problems, and genital abnormalities. When the literature is examined, it is seen that approximately 110 patients have been reported to date.4 In this case report, we aim to present the clinical findings of a seven-year girl who was admitted to the hospital with neuromotor retardation and was diagnosed with KS.
CASE REPORT
A 7-year girl was admitted to the paediatric neurology outpatient clinic with the complaint of aphasia. It was learnt that she had been followed up with neuromotor retardation; and epilepsy was diagnosed at another centre.
Generalised epileptiform activity was observed in her electroencephalography (EEG), taken two years ago, and treatment was started with valproic acid but terminated by the family on the grounds that she did not have seizures as frequently as before while using the drug. While the patient's prenatal and natal history did not reveal any significant findings, it was disclosed that she began to sit at 2 years, walk at 2.5 years and speak one word sentences at 3 years of age. There was no history of consanguinity between her parents, and she received special education due to clinical diagnosis of autism. When the patient's history was further explored, it was revealed that she had generalised tonic-clonic seizures lasting 3-4 minutes once a month. On physical examination, her body weight was 22 kg (25-50 percentile), head circumference was 48.5 cm (<2 SD), and she made no eye contact, and had stereotypic movements, mental retardation, horizontal nystagmus, kyphoscoliosis, coarse facial appearance, hypotonia in the lower extremities, and sacral dermal sinus. Other systemic examinations were normal (Figure 1).
On laboratory examination, hemogram, biochemistry, thyroid function tests (Free T4 and TSH), vitamin B12, folate, 25-OH vitamin D levels, metabolic tests (urine and blood amino acids, tandem MS amino acid and acylcarnitine, urine organic acids, biotinidase, ammonia, and lactate) were normal. On cranial magnetic resonance imaging (MRI), the trunk of corpus callosum and splenium were hypoplasic (Figure 2). Lumbosacral MRI was normal. Sleep EEG was normal. Valproic acid was re-started gradually at two doses of 20 mg/kg/day. Due to abnormal facies and neuromotor retardation, she was referred to the Genetics Department. Molecular karyotyping revealed a loss of 531 kbp covering the 9q34.3 region (Figure 3). After being diagnosed with KS, she was referred to the paediatric cardiology department for associated cardiac pathologies and to the paediatric psychiatry department for associated psychiatric disorders. No pathology was observed on hearing test, ophthalmologic examination and whole abdomen ultrasonography. The patient was followed up for 9 months, and there were no convulsions at her follow-up.
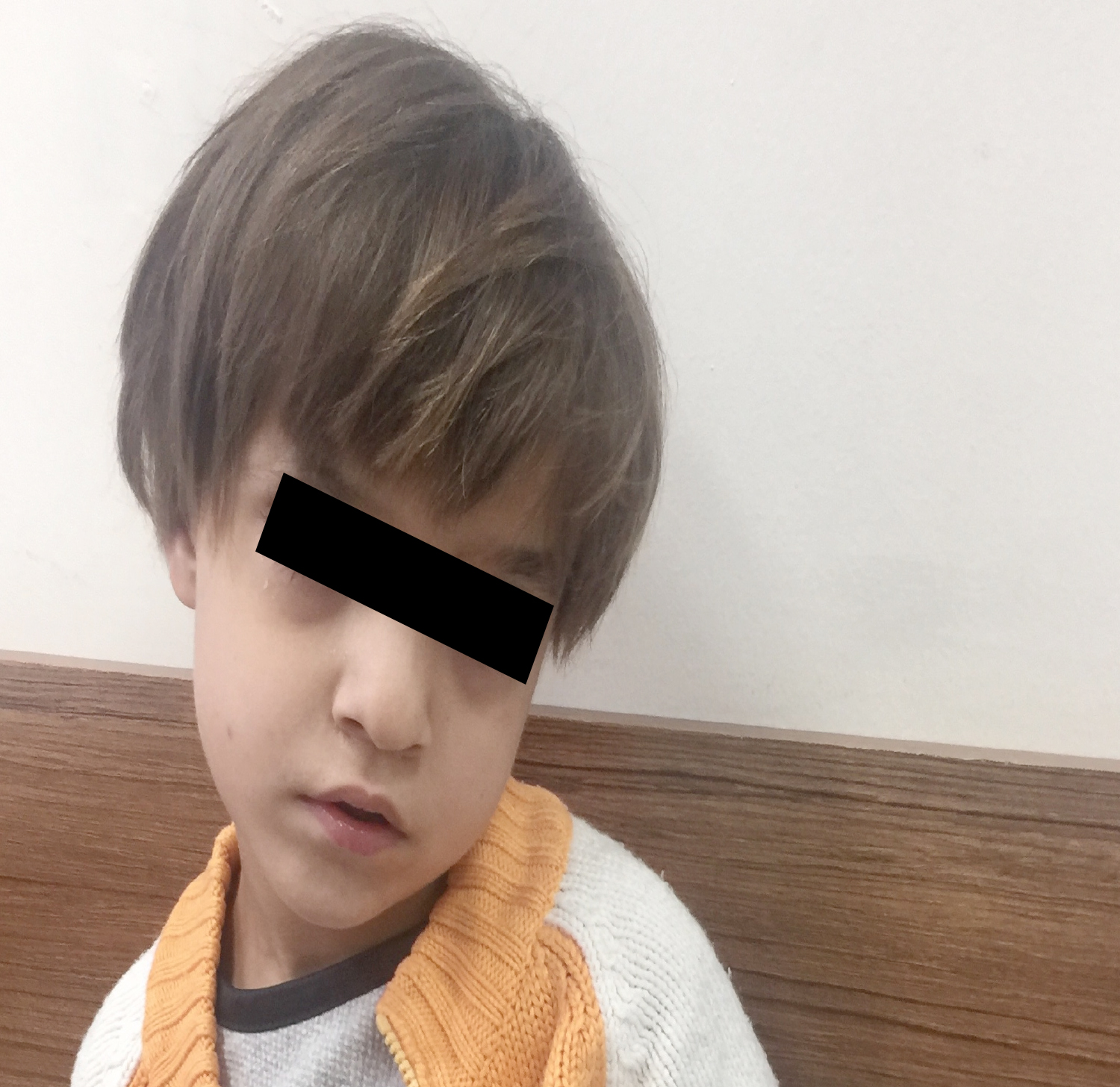 Figure 1: Phenotypic findings of our patient with abnormal course facies.
Figure 1: Phenotypic findings of our patient with abnormal course facies.
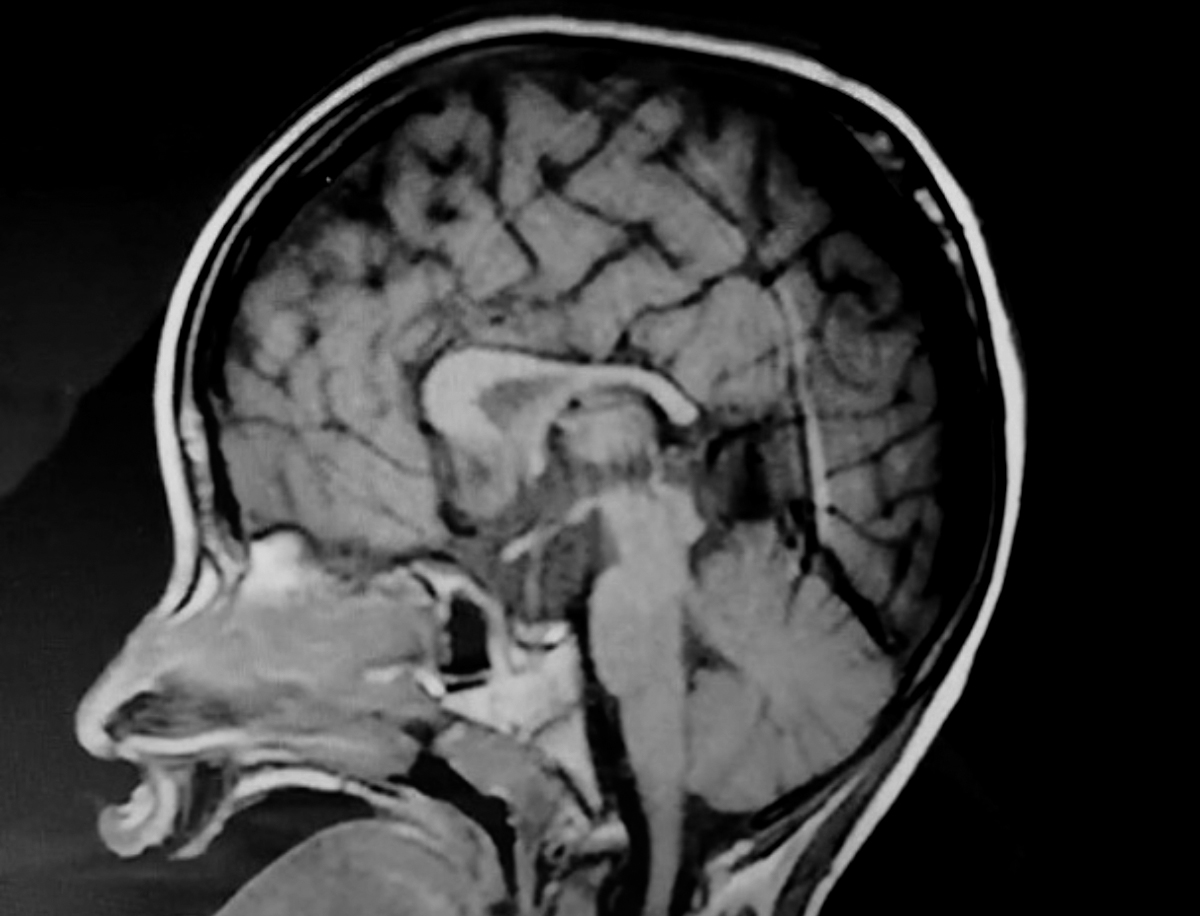 Figure 2: Cranial magnetic resonance imaging (MRI) shows hypoplasia of the trunk of corpus callosum and splenium.
Figure 2: Cranial magnetic resonance imaging (MRI) shows hypoplasia of the trunk of corpus callosum and splenium.
DISCUSSION
KS, previously referred to as the 9q subtelomeric deletion syndrome (9qSTDS), is mainly characterised by moderate to severe developmental delay/mental retardation, infantile hypotonia and brachy-microcephaly, midface hypoplasia, prognathism, shape anomalies of the lips and eyebrows, which are the main clinical phenotypes.5 The case included in this study had mental motor retardation, hypotonia in the lower extremities, microcephaly and coarse facial appearance.
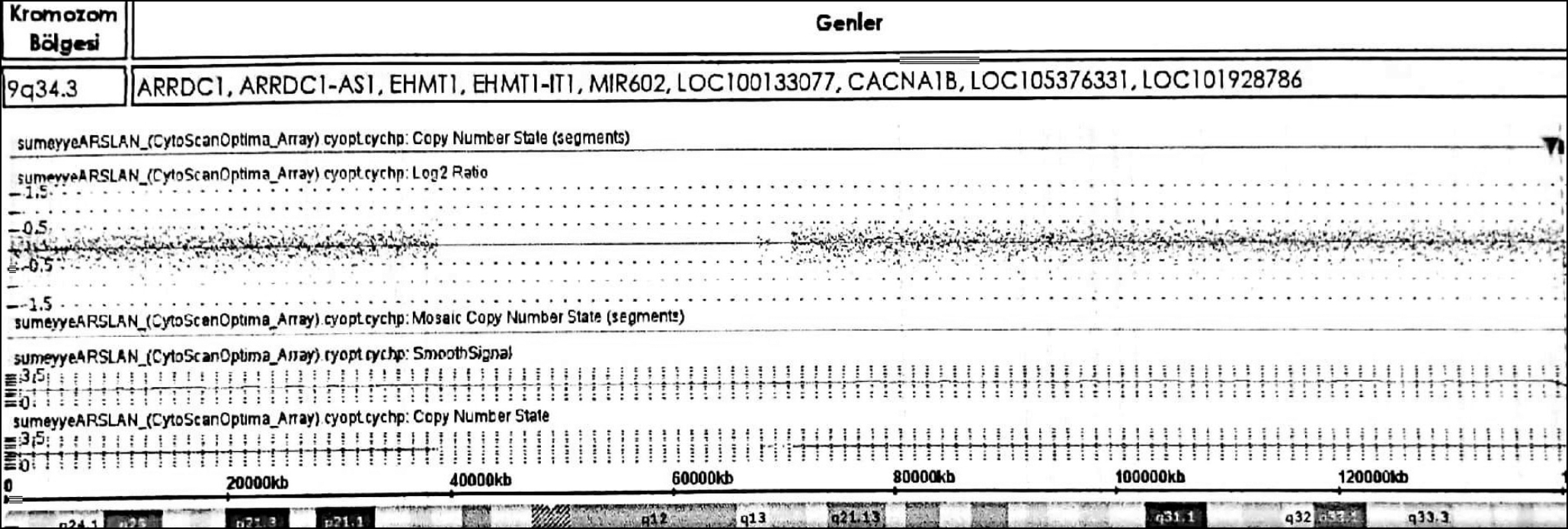 Figure 3: Molecular karyotyping shows a loss of 531 kbp covering the 9q34.3 region.
Figure 3: Molecular karyotyping shows a loss of 531 kbp covering the 9q34.3 region.
Infantile hypotonia is observed in KS; however, almost all cases begin to walk independently after 2-3 years of age. Our case had neuromotor developmental delay, and it was learnt that she began to walk at 2.5 years and sit at 2 years of age. Hearing and visual impairment are very common in this syndrome. Hypermetropia is reported to be seen especially at young ages. Hearing loss (conductive and neural) also occurs in this period.5 Despite the reports in the literature on hearing and vision impairments, ophthalmologic and hearing examinations of our case were normal.
Congenital heart defects such as atrial septal defect (ASD), ventricular septal defect (VSD), tetralogy of Fallot, coarctation of the aorta, bicuspid aortic valve and pulmonary stenosis are observed in patients with KS. Conotruncal heart defects are present in 50% of the cases.4 No structural heart defects were observed on the echocardiography of our case.
Seizures occur in 30% of individuals with KS; tonic-clonic seizures, absence and complex partial seizures can be observed in these patients. Valproic acid was re-started, since our case was found to have a history of seizures, and her EEG findings were consistent with epilepsy. On cranial imaging of cases with KS, the syndrome is accompanied by corpus callosum hypoplasia, dilated ventricles, white matter abnormalities, and cerebellar hypoplasia.6 Cranial imaging of our case also supported the previously reported findings.
KS should be distinguished from other syndromes that include developmental delay, infantile hypotonia, short stature, distinctive facies and behavioural phenotype, such as Down syndrome, Smith-Magenis syndrome, Pitt Hopkins syndrome and Angelman syndrome.
In conclusion, KS is a very rare and genetically diagnosed disease with variable phenotypic findings that may be seen in other hereditary syndromes. Genetic analysis should be planned, and interdisciplinary monitoring should be provided in cases suspected to have KS syndrome.
PATIENT’S CONSENT:
Informed consent was taken from the patient.
CONFLICT OF INTEREST:
Neither of the authors has any conflict to declare.
AUTHORS’ CONTRIBUTION:
HA, IHB: Drafted the case summary and discussion, drafted and edited the manuscript.
HA, IHB, HB: Established and confirmed the diagnosis, drafted the manuscript and references.
REFERENCES
- Stewart DR, Kleefstra T. The chromosome 9q subtelomere deletion syndrome. Am J Med Genet C Semin Med Genet 2007; 145(4):383-92. doi: 10.1002/ajmg.c.30148.
- Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (ehmt1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet 2006; 79(2):370-7. doi: 10.1086/505693.
- Bock I, Németh K, Pentelényi K. Targeted next generation sequencing of a panel of autism-related genes identifies an EHMT1 mutation in a Kleefstra syndrome patient with autism and normal intellectual performance. Gene 2016; 595(2):131-41. doi: 10.1016/j.gene.2016.09.027.
- Verhoeven WM, Egger JI, Vermeulen K, van de Warrenburg BP, Kleefstra T. Kleefstra syndrome in three adult patients: Further delineation of the behavioral and neurological phenotype shows aspects of a neurodegenerative course. Am J Med Genet A 2011; 155(10):2409-15. doi: 10.1002/ ajmg.a.34186.
- Willemsen MH, Vultovan Silfhout AT, Nillesen WM, Wissink-Lindhout WM, Van Bokhoven H, Philip N, et al. Update on kleefstra syndrome. Mol Syndromol 2011; 2(5):202-12. doi: 10.1159/000335648.
- Yatsenko SA, Cheung SW, Scott DA, Nowaczyk MJ, Tarnopolsky M, Naidu S, et al. Deletion 9q34.3 syndrome: Genotype-phenotype correlations and an extended deletion in a patient with features of Opitz C trigonocephaly. J Med Genet 2005; 42(4):328-35. doi: 10.1136/jmg.2004.028258.