First Documented Case of Pulmonary Alveolar Proteinosis with Atopy Presenting Secondary to CSFR2B Mutation
By Ayse Senay Sasihuseyinoglu1, Dilek Ozcan1, Arbil Avci2, Gursu Kiyan3Affiliations
doi: 10.29271/jcpsp.2022.08.S183ABSTRACT
Pulmonary alveolar proteinosis (PAP) is a rare lung disorder in which surfactant-derived lipoproteins accumulate excessively within pulmonary alveoli, causing severe respiratory distress. It is essential to gain a better understanding of the signs to clinically diagnose PAP and include PAP among the differential diagnoses of interstitial pulmonary diseases or other diseases with similar manifestations. We describe a 2.5-year patient with atopy who presented with pulmonary infiltration, recurrent wheezing, and cough despite steroid and salbutamol administration via inhalation. High-resolution computed tomography revealed crazy-paving patterns in both lungs, suggesting PAP. An open lung biopsy revealed intra-alveolar granular amphophilic material, which was strongly positive on periodic acid-Schiff staining. The results of pulmonary-associated surfactant protein B and C gene analyses were normal. However, granulocyte-macrophage colony-stimulating factor receptor beta-protein was not detected in leucocytes, and a novel mutation was identified in the CSF2RB gene. The patient was diagnosed with PAP and treated with whole-lung lavage.
Key Words: Pulmonary alveolar proteinosis, Child, Atopy, Wheezing.
INTRODUCTION
Pulmonary alveolar proteinosis (PAP) is a rare disease that causes respiratory distress due to the accumulation of surfactant lipoproteins in the alveoli.1 PAP is diagnosed based on the presence of homogeneous ground-glass opacities (GGOs) with thickened intralobular and interlobular septa, referred to as ‘crazy-paving’ patterns that can be observed using high-resolution computed tomography (HRCT).2
Atopy is described as a predisposition to immunoglobulin (Ig)E antibody production against allergens that cause asthma, food allergies, rhinoconjunctivitis, and atopic dermatitis. Sensitisation is defined as levels of allergen-specific IgE ≥0.35 kUA/L. However, positive allergy test results do not always imply a clinical allergic reaction on allergen exposure.3
Here, we present the case of a 2.5-year child with atopy who was referred for asthma, but was diagnosed with congenital PAP.
CASE REPORT
A 2.5-year girl was admitted to our clinic with recurrent wheezing, cough, and dyspnoea. She had remained healthy until the age of 2 years when she was hospitalised for bronchiolitis. Her parents were consanguineous, but there was no significant family history.
Physical examination on admission revealed the following results: bodyweight, 8.5 kg (<5th percentile); height, 72 cm (<5th percentile); body temperature, 38.5°C; respiratory rate, 54 breaths/min; peak apical heart rate, 133 beats/min; blood pressure, 90/60 mmHg; and SpO2, 90% of room temperature. Nasal flaring, intercostal retractions, bilateral crackles, and rhonchi were noted on respiratory system examination. The whole blood count, erythrocyte sedimentation rate, biochemical test results, lymphocyte subsets, Ig levels, and echocardiography results were normal. Polymerase chain reaction findings for the respiratory syncytial virus, parainfluenza virus, cytomegalovirus, Epstein–Barr virus, tuberculosis test results, and sweat test results were negative. The Phadiatop-based values of common inhalant allergens were 0.57 pAU/L, 2.66 kAU/L for milk-specific IgE, and 6.2 kAU/L for egg white-specific IgE. However, the results of skin prick and food provocation tests with milk and egg were negative. Therefore, allergic diseases were ruled out. The patient remained symptomatic despite the administration of oxygen, inhaled and systemic steroids, salbutamol, aminophylline, tazocin, and ribavirin. Chest radiography revealed disseminated infiltration in both lungs (Figure 1). Bilateral GGOs were observed on HRCT. Alveoli and terminal bronchioles containing periodic acid–Schiff (PAS)-stained eosinophilic material were noted on histopathological examination, and this finding was compatible with PAP (Figure 2). Granulocyte-macrophage colony-stimulating factor (GM-CSF), GM-CSF receptor alpha and beta-protein, and GM-CSF autoantibody concentration measurements and the signal transducer and activator of transcription 5 (STAT5) stimulation index tests were performed. Her serum GM-CSF concentration measured using an enzyme-linked immunosorbent assay was 70.3 pg/mL (normal = 0–7.5 pg/mL). GM-CSF receptor beta-protein and the STAT5 stimulation index was equal to 0. A novel mutation was reported in the CSF2RB common subunit gene. She was diagnosed with congenital PAP. Therapeutic whole-lung lavage (WLL) was performed. Clinical improvement was then observed, and chest radiography revealed regression in the infiltration (Figure 3).
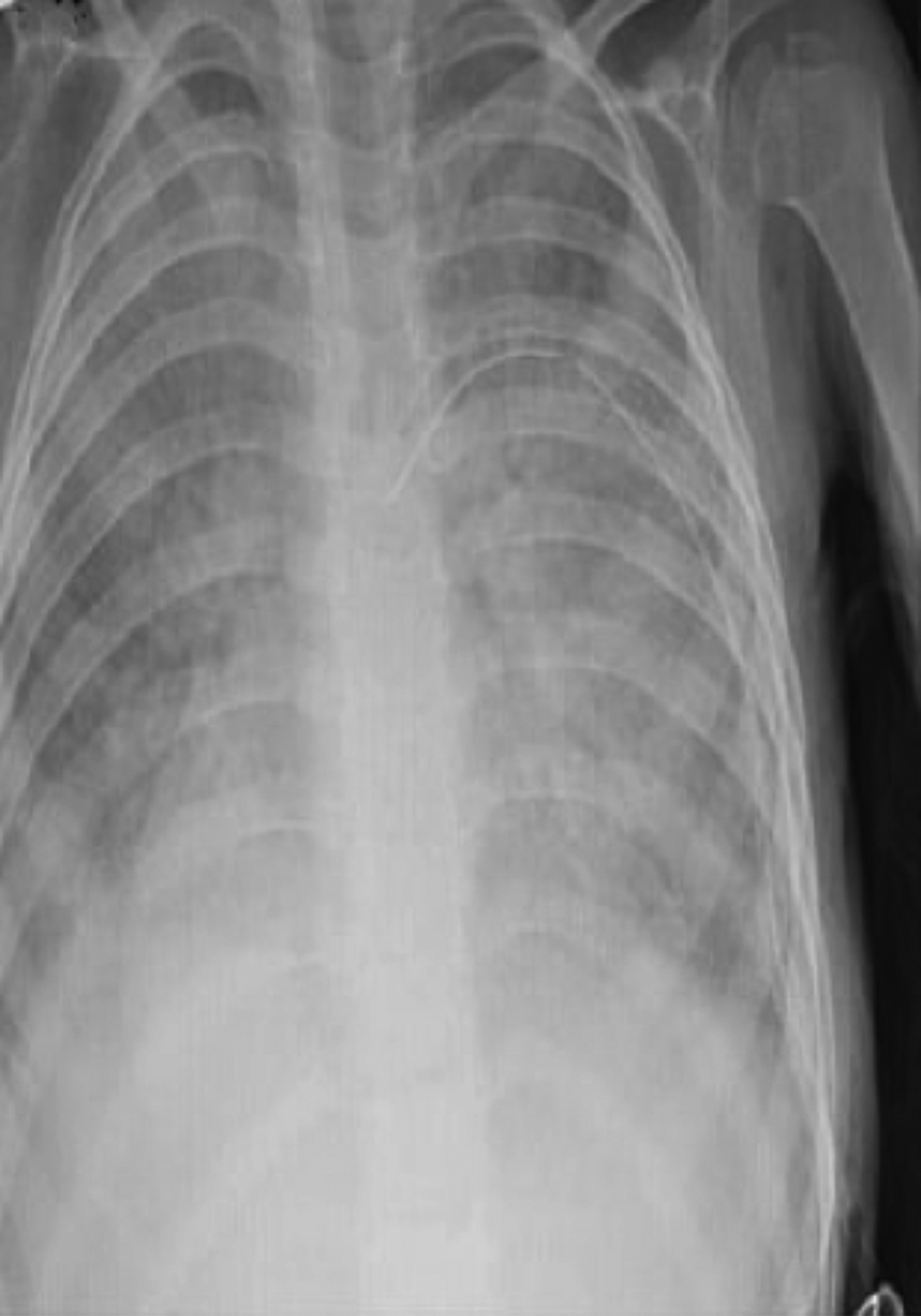 Figure 1: Chest X-ray taken at the time of diagnosis: Disseminated lesions visible in the lung.
Figure 1: Chest X-ray taken at the time of diagnosis: Disseminated lesions visible in the lung.
DISCUSSION
Pulmonary surfactant is composed of phospholipids and surfactant proteins A–D. GM-CSF stimulates the clearance of surfactant by alveolar macrophages through signals transmitted via cell-surface receptors comprised of ligand-binding alpha and affinity-enhancing beta chains.4 Congenital, secondary, and autoimmune or primary are three recognised forms of PAP.5 Anti-GM-CSF antibodies block the activation of alveolar macrophages in the autoimmune form of PAP. Secondary PAP is caused by damage to alveolar macrophages due to inorganic dust, toxic gas inhalation, or infections and haematological malignancies.
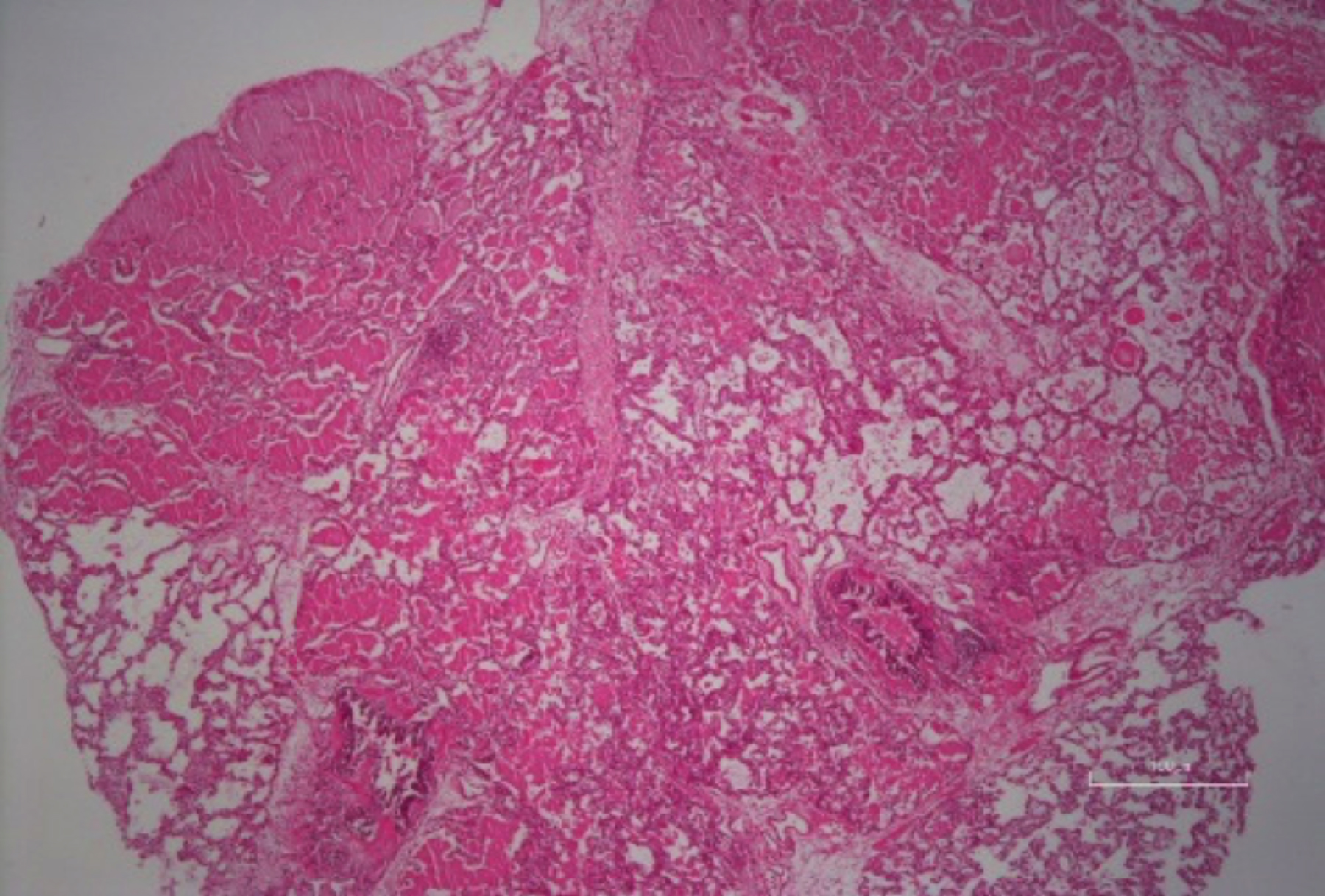 Figure 2: Many alveolar spaces are almost completely filled with homogenous pink staining proteinaceous material (hematoxylin and eosin, ×100).
Figure 2: Many alveolar spaces are almost completely filled with homogenous pink staining proteinaceous material (hematoxylin and eosin, ×100).
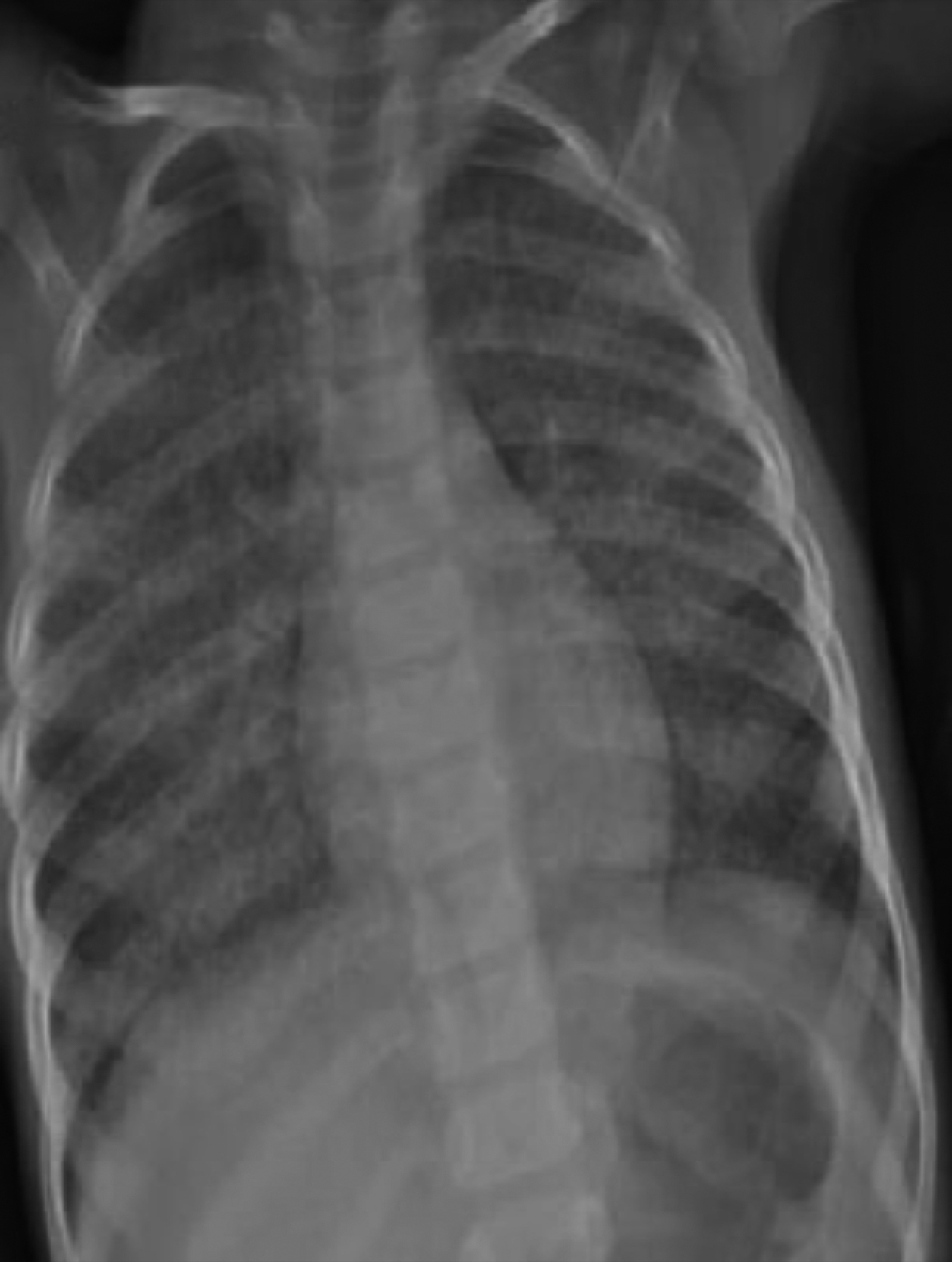 Figure 3: Chest X-ray taken after whole-lung lavage.
Figure 3: Chest X-ray taken after whole-lung lavage.
No such organic pathology was detected in this case, and the serum GM-CSF autoantibody test results were negative. Congenital PAP is caused by mutations in genes encoding surfactant proteins, ABCA3, or chains in GM-CSF receptors.5 This patient had a high GM-CSF concentration, lacked GM-CSF receptor beta-protein, and was diagnosed with congenital PAP.
The common clinical manifestations of PAP are progressive dyspnoea, dry cough, fatigue, and low-grade fever. These symptoms are observed in atopic conditions. However, the pathophysiology is completely different. In patients with PAP, areas of bilateral and symmetrical alveolar opacities are usually observed on chest radiography. An important diagnostic tool is HRCT, which usually detects lesions such as GGOs, including the ‘crazy paving’ pattern.6 Characteristic bronchoalveolar lavage findings corresponding to PAP include an opaque or milky appearance of the fluid which settles on standing. Lung biopsy shows a preserved alveolar architecture, slight thickening of the alveolar septa, little or no inflammatory cell infiltration, and the presence of PAS-positive proteinaceous material with a background of eosinophilic granules in terminal bronchioles and alveoli.
Thus, the most appropriate and safe treatment for PAP to clear alveoli is WLL.6
To the best of our knowledge, this patient represents the first documented case of PAP with atopy. This case emphasises that in patients presenting with unexplained recurrent wheezing, cough, and dyspnoea with radiological findings of bilateral diffuse infiltration, physicians should consider PAP among the differential diagnoses. Positive allergy test results do not always indicate the presence of atopic disease.
COMPETING INTEREST:
The authors declared no competing interest.
PATIENT’S CONSENT:
Written informed consent for publication was obtained from the parents of the patient.
AUTHORS’ CONTRIBUTION:
ASS: Conception, design, literature search, and manuscript writing.
AA, DD: Interpretation of data and revising.
GK: Interpretation of data, revising and supervision of the final version english redaction.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Yoon HY, Kim JH, Kim YJ, Song JW. Pulmonary alveolar proteinosis in Korea: Analysis of prevalence and incidence via a nationwide population-based study. BMC Pulm Med 2020; 20(1):34. doi: 10. 1186/s12890-020-1074-5.
- Allwood BW, Bennji S. Crazy paving in pulmonary alveolar proteinosis. N Engl J Med 2020; 382(3):275. doi: 10. 1056/NEJMicm1908563.
- Comberiati P, Di Cicco ME, D'Elios S, Peroni DG. How much asthma ıs atopic in children? Front Pediatr 2017; 5:122. doi: 10.3389/fped.2017.00122.
- Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, et al. The granulocyte-macrophage colony-stimulating factor receptor: Linking its structure to cell signalling and its role in disease. Blood 2009; 114(7): 1289-98. doi: 10.1182/blood-2008-12-164004.
- Hadda V, Tiwari P, Madan K, Mohan A, Gupta N, Bharti SJ, et al. Pulmonary alveolar proteinosis: Experience from a tertiary care center and systematic review of Indian literature. Lung India 2016; 33(6):626-34. doi: 10.4103/ 0970-2113.192876.
- Griese M. Pulmonary alveolar proteinosis: A comprehensive clinical perspective. Pediatrics 2017; 140(2):e20170610. doi: 10.1542/peds.2017-0610.