A Rare Case of Propionic Acidemia in a Six Months Female Child
By Sobia Irum, Ambreen Rehman, Muhammad Aamir, Zujaja Hina Haroon, Nayyar Chaudhry, Afshan BibiAffiliations
doi: 10.29271/jcpsp.2022.08.S180ABSTRACT
Propionic Acidemia (PA) is a rare metabolic disorder caused by the defect in enzyme (propionyl-coenzyme A (CoA) carboxylase) leading to the abnormal accumulation of metabolites of branched-chain amino acid catabolism in blood and urine. We describe the first ever diagnosed case in our setup of early onset PA in a 06 months old baby girl who presented with the complaints of decreased feed intake, lethargy, vomiting, failure to thrive, and intermittent seizures. The basic laboratory investigations showed pancytopenia along with high anion gap metabolic acidosis, urine dipstick positive for ketones, and hyperammonemia. Plasma amino acid analysis by ion exchange chromatography (IEC) showed elevated plasma glycine and lysine levels. Finally, urine organic acid analysis by gas chromatography-mass spectrometry (GCMS) showed marked elevation of 3-hydroxy propionate, methyl citrate, and 3-hydroxy, 2 methylbutyric acid with moderate rise in 3-hydroxy butyric acid without an elevation of methylmalonate in urine, thus giving the diagnosis of PA.
Key Words: Propionic acidemia, Propionyl-CoA Carboxylase deficiency, Gas chromatography-mass spectrometry.
INTRODUCTION
Propionic acidemia (PA) is a rare autosomal recessive and inherited metabolic disorder with overall incidence of 1: 100,000 to 150,000 and is caused by the defective form of mitochondrial enzyme (propionyl-coenzyme A (CoA) carboxylase) which results in the accumulation of propionic acid. Propionyl-CoA carboxylase converts propionyl-CoA to methylmalonyl-CoA. Several genetic mutations, broadly categorised as defects in 2 sub-units of the propionyl-CoA carboxylase gene (PCCA and PCCB), may give rise to varying levels of functioning propionyl-CoA carboxylase. Defects in the metabolic pathway produce several potentially toxic metabolites.1 Classical neonatal PA symptoms can occur within the days of birth. These can include poor feeding, weight loss, vomiting, lethargy, neurological involvement, metabolic acidosis, and death. PA can manifest at any age depending partly on the severity of mutations in PCCA and PCCB genes and on environmental inputs.2 The common laboratory abnormalities during acute decompensation include high-anion gap metabolic acidosis, lactic acidosis, elevated plasma and urinary ketones, low to normal blood glucose concentration, hyperammonemia, neutropenia, anemia, and thrombocytopenia.
Since the clinical presentations of PA are non-specific, misdiagnosis leading to the mistreatment or inadequate treatment causes deterioration of metabolic decompensation and, if left untreated, lead to the death.3 Timely diagnosis and treatment can lead to an improved quality and quantity of life. The diagnostic facilities / investigations are available in the form of urinary organic acids by gas chromatography-mass spectrometry (GCMS)4 which shows high concentrations of metabolites of propionyl-CoA including propionic acid, methyl citrate, 3-OH propionic acid, 3-OH 2 methyl butyric acid, tiglic acid, tiglylglycine, and propionylglycine, and quantitative measurements of amino acids in plasma and urine which shows increased glycine and lysine concentrations. Confirmation is done with molecular genetic testing which reveals a pathogenic variant in PCCA or PCCB or deficient propionyl-CoA carboxylase enzyme activity.5 The disease process can be contained by timely management which includes supporting normal development and quality of life along with the prevention of the episodes of metabolic decompensation and complications. A protein-restricted diet (1.5-2 mg/kg/day) along with supplementation of L-carnitine (100 mg/kg/day), and biotin (10 mg/day) is the cornerstone of the treatment.6
CASE REPORT
A 06-months female child was referred to the Pediatric Metabolic Laboratory for urine organic acid and plasma amino acid analysis for evaluation of inborn metabolic disorder. She presented with the complaints of decreased feed intake, lethargy, vomiting, constipation, failure to thrive, and intermittent seizures for the last 3 months. This female infant had birth weight of 3.1 Kg and was delivered at term with the normal Apgar scores to a second para mother through consanguineous marriage following an uneventful antenatal period. She was taking formula milk. She had a history of meningitis at 3 months of age. The general physical examination showed a pale lethargic child with moderate to severe dehydration. Her weight on presentation was 4 Kg and occipitofrontal circumference (OFC) was 38 cm. On CNS examination, there was hypotonia and hyporeflexia while the rest of systemic examination was unremarkable.
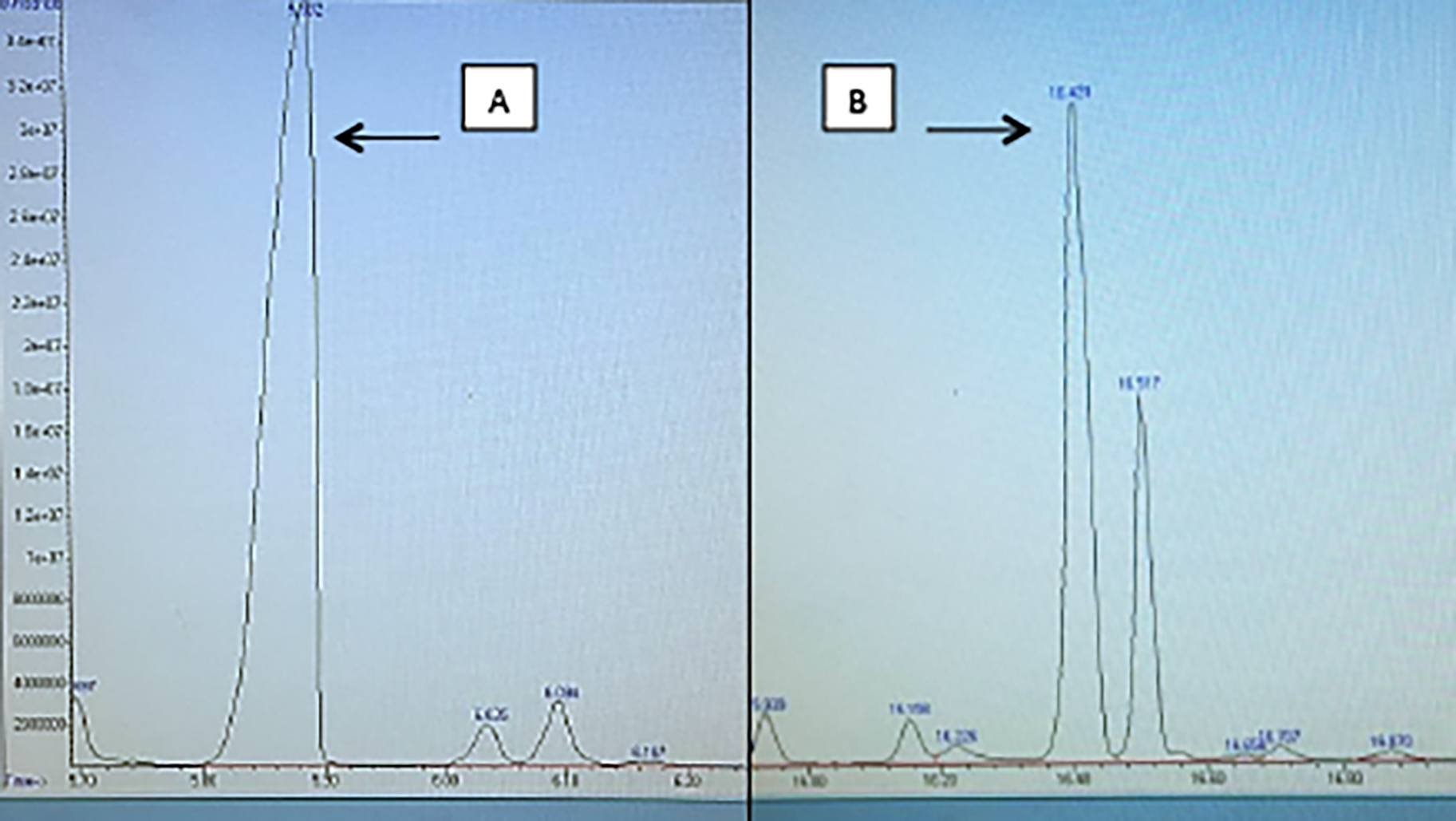 Figure 1 (a, b): Urine organic acid chromatogram showing markedly increased peaks of 3-hydroxy propionic acid and methyl citrate, respectively.
Figure 1 (a, b): Urine organic acid chromatogram showing markedly increased peaks of 3-hydroxy propionic acid and methyl citrate, respectively.
The biochemical findings revealed: (1) Blood screening tests showed high anion gap metabolic acidosis, urine dipstick was positive for ketones, and raised plasma ammonia. Blood complete picture showed pancytopenia with normal lactate and blood glucose. (2) Urine organic acid analysis by GC MS showed marked elevation of 3 hydroxy propionate (1447 µmol/mmol creatinine; Reference range: <26 µmol/mmol creatinine), methyl citrate (684 µmol/mmol creatinine; Reference range: <8 µmol/mmol creatinine), and 3-hydroxy 2 methylbutyric acid with moderate rise in 3-hydroxy butyric acid without an elevation of methylmalonate. 3 Plasma amino acid analysis in lithium heparin tube by ion exchange chromatography (IEC) showed elevated glycine of 1067 µmol/L (Reference range: 107-343 µmol/L) and elevated lysine value of 516 µmol/L (Reference range: 66-270 µmol/L). The lumbar tap was normal, and blood culture was sterile which ruled out meningitis.
Based on history, clinical examination, and above-mentioned laboratory findings, the case was labelled as PA. As enzyme assays were not available, exact enzyme deficit could not be confirmed. Acidosis was appropriately and adequately treated with sodium bicarbonate. The patient was started on Cap Biotin 10 mg once a day, Tab L-Carnitine 400 mg once a day, and a protein deficient formula for feeding.
Her clinical condition improved. Currently, she is being followed up on fortnightly basis for checking any improvement in her biochemical profile and mental activity with the diet modification and supplementation.
DISCUSSION
This case is the first ever diagnosed and reported in paediatric metabolic laboratory in Pakistan. The diagnosis was confirmed by urine organic acids analysis which showed marked elevation of 3 hydroxy propionate, methyl citrate, and 3-hydroxy 2 methyl butyric acid with moderate rise in 3-hydroxy butyric acid without an elevation of methylmalonate. PA, due to deficiency of the enzyme propionyl-coenzyme A (CoA) carboxylase, is a rare inherited metabolic disorder with overall incidence of 1: 100,000 to 150,000.7 Our case presented with non-specific symptoms of decreased feed intake, lethargy, vomiting, constipation, failure to thrive, and intermittent seizures, which are also seen in other metabolic and non-metabolic disorders like sepsis, gastrointestinal obstruction, cardiorespiratory difficulties, or birth trauma but these symptoms are also seen in typical case of PA.7 In this case, CNS findings of hypotonia and hyporeflexia were present as in many other documented cases.7 Cardiomyopathy was not present in this case as it was seen in approximately 50% of PA cases.8 In this case, magnetic resonance imaging of the brain showed normal study due to the early presentation.
The deficiency of propionyl-coenzyme A (CoA) carboxylase leads to abnormal accumulation of metabolites of branched-chain amino acid catabolism in blood and urine. These metabolic findings were also seen in this patient like, marked elevation of 3 hydroxy propionate, methyl citrate, 3-hydroxy 2 methyl butyric acid, and moderate rise in 3-hydroxy butyric acid on urine organic acid analysis along with elevated glycine and lysine levels on plasma amino acid analysis. These findings are diagnostic of PA, which is mentioned in recommended guidelines.7 Preliminary investigations like pancytopenia, high anion gap metabolic acidosis, and raised ammonia with positive urinary ketones further reinforced this diagnosis.
The confirmation of PA should ideally be done by molecular genetic testing of the pathogenic variants in PCCA or PCCB genes or by the measurement of propionyl-CoA carboxylase enzyme activity. It was, however, not carried out in this patient due to non-availability of the facility in our set-up.
The aim of the treatment is to prevent endogenous protein catabolism whilst providing enough energy to promote anabolism by giving low-protein diet, L-carnitine supplementation, and biotin supplementation.
PATIENT’S CONSENT:
Informed consent has been obtained from patient’s father to publish the data concerning this case.
COMPETING INTEREST:
All the authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
SI: Contributed to drafting and preparation of report.
AR: Contributed to interpretation and preparation of report.
MA: Final approval of case report for submission.
ZHH: Revising critically for any intellectual content.
NC: Revising for accuracy and integrity of all the parts.
AB: Critical revision of the content.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Almási T, Guey LT, Lukacs C, Csetneki K, Vokó Z, Zelei T. Systematic literature review and meta-analysis on the epidemiology of propionic acidemia. Orphanet J Rare Dis 2019; 14(1):40. doi: 10.1186/s13023-018-0987-z.
- Barry MA. Recent advances towards gene therapy for propionic acidemia: translation to the clinic. Expert Rev Precis Med Drug Dev 2019; 4(4):229-37. doi: 10.1080/2380 8993.2019.1635883.
- Wongkittichote P, Mew NA, Chapman KA. Propionyl-CoA carboxylase – a review. Mol Genet Metab 2017; 122(4): 145-52. doi: 10.3389/fcvm.2020.617451.
- Wajner M, Sitta A, Kayser A, Deon M, Groehs AC, Coelho DM, et al. Screening for organic acidurias and amino-acidopathies in high-risk Brazilian patients: Eleven-year experience of a reference center. Genet Mol Biol 2019; 42(1):178-85. https://doi.org/10.1590/1678-4685-GMB-20 18-0105.
- Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014; 9(1):130. doi.org/10. 1186/s13023-014-0130-8.
- Burlina A, Tims S, van Spronsen F, Sperl W, Burlina AP, Kuhn M, et al. The potential role of gut microbiota and its modulators in the management of propionic and methylmalonic acidemia. Expert Opin Orphan Drugs 2018; 6(11):683-92. doi.org/10.1080/21678707.2018.1536540.
- Baumgartner MR, Horster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014; 9(1):130. doi.org/10. 1186/s13023-014-0130-8.
- Pena L, Burton BK. Survey of health status and complications among propionic acidemia patients. Am J Med Genet A 2012; 158(7):1641-6. doi.org/10.1002/ ajmg.a.35387.