A Novel Variant in Paired Box 3 Gene-related with Waardenburg Syndrome Type-1 in an Afghan Family
By Mustafa Dogan1, Recep Eroz2Affiliations
doi: 10.29271/jcpsp.2022.08.S110ABSTRACT
Waardenburg Syndrome (WS) is a congenital auditory-pigmentary syndrome. We, herein, present a case of a 1.5 year girl presenting with bilateral hearing impairment. Detailed examinations and molecular analyses of the proband and other family members were performed. A novel missense, heterozygous variant (c.253A>C (p.Lys85Gln)) was detected in the paıred box 3 (PAX3) gene. For interpretation and classification of the variant, the American College of Medical Genetics and Genomics (ACMG) guideline was used. No previous report of this variant was found in the literature and we determined the variant according to the guide published in 2015 as ‘’likely pathogenic’’. We think that the clinical and genetic characterisation of the current family will contribute to knowledge for a better understanding of the genetic background of the Afghan patients with WS.
Key Words: Waardenburg syndrome, Congenital auditory-pigmentary syndrome, PAX3 gene.
INTRODUCTION
Waardenburg Syndrome (WS) is a neurocristopathy with an autosomal dominant mode of inheritance but has also been seen in recessive cases1 and is a heterogeneous disease. Until now, mutations of the PAX3, SOX10, MITF, EDNRB, EDN3, and SNAI2 genes have been shown to play a role in the etiopathogenesis of WS and the disease is one of the most common causes of syndromic hearing loss with variable expressivity. The prevalence of WS is estimated from 1:20,000 to 1:42,000 and is present in nearly 5% of the patients with congenital deafness.2 The characteristic features of the disease are sensorineural hearing impairment, pigmentation abnormalities such as depigmented patches of the skin and hair, heterochromia iridis, and dystopia canthorum (lateral displacement of the inner canthi), musculoskeletal defects, or Hirschsprung’s disease.2,3
In this report, we present a case of a rare non-consanguineous Afghan family with WS type 1 with a novel heterozygous variant in the PAX3 gene.
CASE REPORT
The proband is a 1.5 year girl with bilateral hearing impairment referred to the medical genetics clinic for genetic testing. Individual’s informed consent was obtained from the parents for the molecular studies and case reports. The color of the skin, hair, iris, and the presence of developmental defects were evaluated. According to the history taken from the mother, her prenatal history was unremarkable and no pregnancy complications were reported. She was the third child of non-consanguineous parents of Afghan descent and her elder sister and brother were phenotypically normal. She had iris heterochromia in both of the right and left eyes, dystopia canthorum, mild synophrys, and broad nasal root but no pigment abnormalities of the hair and skin (Figure 1). Neuromotor retardation was identified, but there were no neurological defects such as peripheral neuropathy, cerebellar ataxia or spasticity. It was observed that there was no speech in the patient. Musculoskeletal anomalies and Hirschsprung’s disease were absent. Ophthalmologic assessments revealed normal vision. The refugee family stated that newborn hearing screening could not be performed at birth. A hearing aid was given for language rehabilitation during check-ups performed in Turkey but because of the failure of hearing aids to compensate for the profound deafness, cochlear implantation was arranged for the patient. Temporal computed tomography findings were normal. She had been examined in various medical departments, but a definite diagnosis was not established. No cases with similar features were described in her family history while her mother was found to have moderate hearing loss and a white forelock.
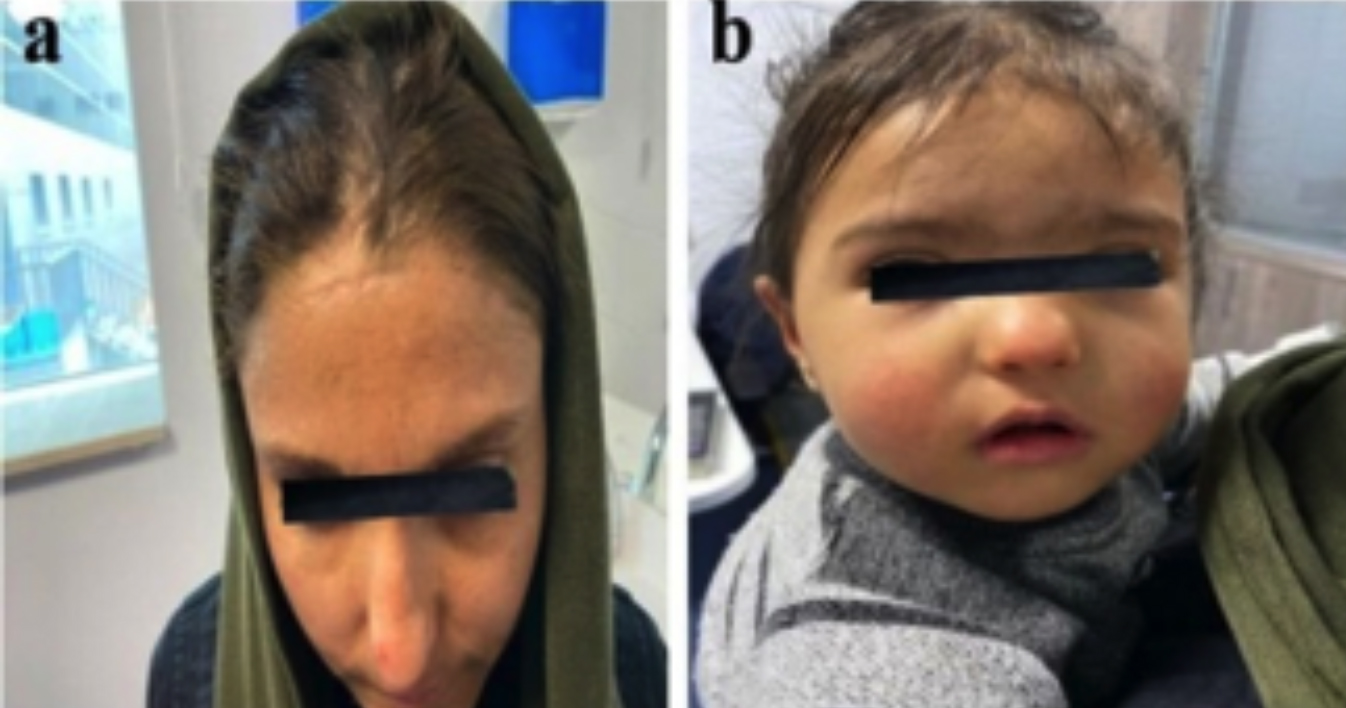 Figure 1: Pictures of the affected individuals.
Figure 1: Pictures of the affected individuals.
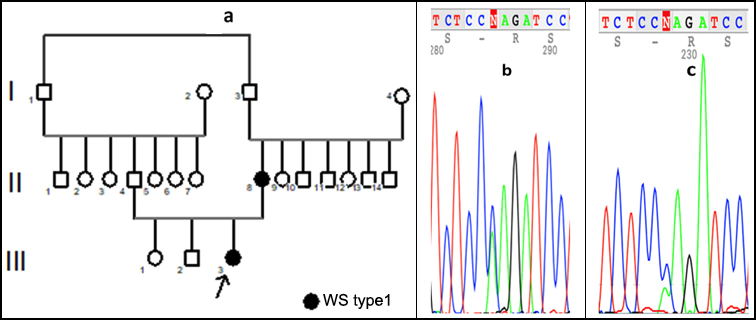 Figure 2: Family pedigree and sequencing view of the variants.
Figure 2: Family pedigree and sequencing view of the variants.
The clinical features of the patient in this study supported the diagnosis of WS type 1. Genomic DNA was isolated from the whole blood sample obtained from the patient. After karyotype analysis was revealed normal, the full coding of the exons, the exon–intron boundaries of the PAX3 gene were performed by using Sanger Sequencing in the index patient and a novel missense, a heterozygous variant (c.253A>C (p.Lys85Gln)) which could explain the patient’s phenotype was detected. The same variation was detected as heterozygous in the patient's mother with a hearing loss problem but neither in the father nor other two siblings. Family pedigree (Figure 2a) and sequencing view of the variant in the PAX3 gene are given in Figure 2 b and c (b: proband and c: mother). We determined the variant according to the guide of ACMG published in 2015 as "likely pathogenic".4 The pathogenicity data for the variant is shown in Table I.
Table I: The pathogenicity data of the variant.
|
GENE |
PAX3 |
|
Ensembl transcript ID |
ENST00000392069.2 |
|
Genbank transcript ID |
NM_181459.4 |
|
Chromosomal locus |
2q36.1 |
|
dbSNP |
novel |
|
Variant |
c.253A>C(p.Lys85Gln) |
|
Variant location |
Exon 2 |
|
Variant type |
Missense |
|
Mutation taster |
Disease causing |
|
SIFT |
Damaging |
|
Provean |
Damaging |
|
ExAC (allele frequency) |
Not found |
|
GnomAD exomes |
No entry |
|
ClinVAR |
- |
|
Conservation |
conserved |
|
DANN score |
0,9946 |
|
ACMG classification |
Likely Pathogenic |
|
ACMG pathogenity criteria |
PM1,PM2,PM5,PP2,PP3 |
DISCUSSION
We presented a small Afghan family with WS type 1 and a novel missense heterozygous variant in the PAX3 gene. The PAX3 gene, located on chromosome 2q36 region, includes 10 exons and encodes a Paired box 3 transcription factor which is involved in neural crest cell border induction at the neural plate. The function of PAX3 is essential for the neural crest from which melanocytes take origin. Apart from hypopigmentation in PAX3 dysfunction, hearing loss occurs due to the absence of melanocytes in the stria vascularis of the inner ear.5 Auditory function varies from normal to profound deafness within the families. Hearing loss, detected in about 60% of the individuals with WS type 1, is congenital, typically non-progressive, can be unilateral or bilateral, and sensorineural type. Bilateral deafness is much more common than unilateral.6 It was noted that the hearing loss in the proband was congenital, sensorineural, and bilateral, but the hearing loss in the mother was moderate. Dystopia canthorum is the most penetrating feature of WS type 1 and this finding was present in both of the studied patients. WS shows a high degree of genetic heterogeneity and no phenotype-genotype correlations within WS type 1 families. However, even among different the patients in the same family, different clinical manifestations may occur due to the differences in the penetration of causal genes. This should be taken into account in genetic counseling.7 Pathogenic variants in the PAX3 gene have been reported in 33-80 % of familial and sporadic cases in patients with WS type 1.8 More than 100 pathogenic variants associated with WS have been identified in the PAX3 gene so far. About half of the mutations detected in the PAX3 gene are missense, and the others are truncating variations.9 With next generation sequencing technology, more variants in the PAX3 gene that causes WS type 1 are detected. The absence of a pathogenic variation in the PAX3 gene of a case with suspected WS type 1 suggests that there may be mutations in other WS or in other genes that have not yet been identified.10
Careful clinical evaluation is required to differentiate WS types. With this study, we present the clinical and genetic findings of an Afghan family with WS type 1. To the best of our knowledge, this case is the first molecularly confirmed WS type 1 patient in the Afghan population in the literature. We detected a novel ‘’likely pathogenic’’ variant (c.253A>C(p.Lys85Gln) in the exon 2 of the PAX3 gene. We think that the current report will contribute to the crucial knowledge for a better understanding of the genetic background of Afghan WS patients. As the number of patients with identified phenotype and genotype correlation in Afghan WS patients increases, there will be more information to obtain.
PATIENT’S CONSENT:
Written informed consents for medical examinations and genomic analyses were obtained from the patient’s parents.
COMPETING INTEREST:
The authors declared no conflict of interest.
AUTHORS’ CONTRIBUTION:
MD: Participated in data acquisition, writing, and critical revision of the manuscript.
RE: Participated in the study concept and supervision of the study.
All the authors have approved the final version of the manuscript to be published.
REFERENCES
- Wang L, Qin L, Li T, Liu H, Ma L, Li W, et al. Prenatal diagnosis and genetic counseling for waardenburg syndrome type I and II in Chinese families. Mol Med Rep 2018; 17(1):172-8. doi: 10.3892/mmr.2017.7874
- Zaman A, Capper R, Baddoo W. Waardenburg syndrome: More common than you think. Clin Otolaryngol 2015; 40(1):44-8. doi: 10.1111/coa.12312
- Gowda VK, Srinivas S, Srinivasan VM. Waardenburg syndrome Type I. Indian J Pediatr 2020; 87(3):244. doi: 10.1007/s12098-019-03170-5.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015; 17(5):405-24. doi: 10.1038/gim.2015.30
- Zhijie N, Jiada L, Fen T, Sun J, Wang X, Jiang L, et al. Identification and functional analysis of a novel mutation in the PAX3 gene associated with Waardenburg syndrome type I. Gene 2018; 642:362-66. doi: 10.1016/j.gene.2017.11.035.
- Oysu C, Baserer N, Tinaz M. Audiometric manifestations of Waardenburg’s syndrome. Ear Nose Throat J 2000; 799(9): 704-9.
- Jing M, Ken L, Hong‐chao J, Yang Y, Zhang Y, Yang G, et al. A novel mutation of the PAX3 gene in a Chinese family with Waardenburg syndrome type I. Mol Genet Genomic Med 2019; 7(7):e798. doi: 10.1002/mgg3.798.
- Ghosh SK, Bandyopadhyay D, Ghosh A, Kumar Biswas S, Kumar Mandal R. Waardenburg syndrome: A report of three cases. Indian J Dermatol Venereol Leprol 2010; 76(5):550-2. doi: 10.4103/0378-6323.69089.
- Milunsky JM, Maher TA, Ito M, Milunsky A. The value of MLPA in Waardenburg syndrome. Genet Test 2007; 11(2):179-82. doi: 10.1089/gte.2006.0531.
- Pingault V, Ente D, Dastot-Le MF, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat 2010; 31(4):391-406. doi: 10.1002/humu.21211.