Hyperargininemic Encephalopathy with Unique Clinical Presentation and Novel Genetic Mutations
By Sundarachary Nagarjunakonda, Rajeswari Daggumati, Sridhar AmalakantiAffiliations
doi: 10.29271/jcpsp.2020.05.535ABSTRACT
Hyperargininemia is a urea cycle disorder that has rarely been reported in adults. We present a case of arginase deficiency disorder in a 32-year man with metabolic encephalopathy. He presented with progressive limb spasticity, changes in personality, cognitive decline (impaired judgement, executive and language dysfunction) and pseudo-bulbar affect. He deteriorated to an akinetic mute and rigid state. MRI brain was suggestive of a metabolic disorder. Hyperammonemia was present, blood arginine levels were elevated, and serum arginase levels were reduced. The standard argI gene mutations were absent but rs2781666 (G/T) and rs2608897 (C/T) variations were noted in this patient.
Hyperargininemic encephalopathy may present in adults and with atypical features. It should be kept in the differential diagnosis of metabolic encephalopathy in adults.
Key Words: Metabolic encephalopathy, Pseudobulbar affect, Arginase deficiency, Hyperammonemia, Urea cycle.
Urea cycle disorders [UCDs] are a group of inherited diseases.1 Hyperargininemia is a type of UCD caused by deficiency of arginase enzyme. The increased arginine and its metabolites like guanidino compounds and nitric oxide are neurotoxic.2 The disorder presents typically in a child as slow, onset of growth restriction, spastic paraplegia, intellectual deficiency and epilepsy. Sometimes ataxia and dystonia might be seen.3 Here, we present a case of hyperargininemia with novel clinical features and genetic variations, in an adult patient.
CASE REPORT
A 32-year hypertensive man was brought to our department with spastic quadriparesis, bowel and bladder incontinence, dysphagia, dystonia of right lower limb, apathy, irritability and floccillation, which progressed to akinetic mutism over 7 months. He had a preference for vegetarian and carbohydrate-rich diet and had a tendency to avoid pulses. He was not a smoker but was habituated to alcohol and had started taking high quantities of alcohol in the preceding four months of the illness (90 ml daily).
Complete blood counts, liver function tests, renal function tests, serum electrolytes, cerebrospinal fluid analysis, arterial blood gas analysis and blood sugar were normal. He was negative for viral markers: human immunodeficiency virus (HIV), hepatitis B surface antigen (HBsAg) and hepatitis C virus (HCV). Serum ammonia was elevated to 82 µmol/L.
MRI brain showed hyper-intense signals on T2 and FLAIR sequences in bilateral fronto-parietal periventricular white matter and in the anterior and posterior limb of internal capsules bilaterally. Hyperintensities on T1 were noted in bilateral putamen, suggestive of hepatic encephalopathy (Figure 1). A search for the causes of a chronic mild hyperammonemic state with normal liver function tests and no other evidence of hepatic encephalopathy was made; and the possibility of late-onset UCD was considered. Biochemical analysis revealed high serum arginine 3283.86, µmol/L [normal: 15 – 128] and low serum arginase 18.52 U/g Hb [normal: 50 – 90] (Table I).
Table I: Metabolic parameters.
|
Investigations |
Values |
Normal range |
|
Ceruloplasmin |
0.38 gm/L |
0.2 – 0.6 gm/L |
|
Copper |
76 µg/dl |
70 – 140 µg/dl |
|
Lactate |
9.8 mg/dl |
4.5 – 20 mg/dl |
|
Ammonia |
82 µmol/L |
11 – 32 µmol/L |
|
Glutamine |
17.89 µmol/L |
205 – 756 µmol/L |
|
Ornithine |
2.88 µmol/L |
48 – 95 µmol/L |
|
Citruline |
0.34 µmol/L |
12 – 55 µmol/L |
|
Arginine |
3283.86 µmol/L |
15 – 128 µmol/L |
|
Arginosuccinic acid |
0.40 µmol/L |
11 – 32 µmol/L |
|
Arginase |
18.52 U/g Hb |
50 – 90 U/g Hb |
Genotyping of the arginase-1 gene of the patient, his 3 family members and one unrelated person (control) was done. Comparison with published arginase mutations found no match with any specific mutation. Two variations in the promoter regions, rs2781666 (G/T) and rs2608897 (C/T), were observed in the patient and two of his family members. These were not found in the control sample.
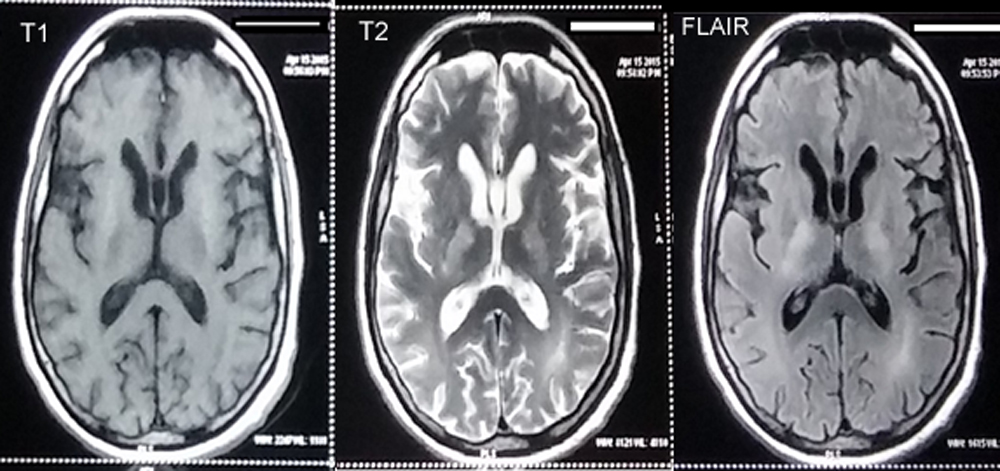 Figure 1: Hyper-intense signals on T2 and FLAIR sequences noted in bilateral fronto-parietal periventricular white matter and in the anterior and posterior limb of internal capsules bilaterally. Hyperintensities on T1 is noted in bilateral putamen.
Figure 1: Hyper-intense signals on T2 and FLAIR sequences noted in bilateral fronto-parietal periventricular white matter and in the anterior and posterior limb of internal capsules bilaterally. Hyperintensities on T1 is noted in bilateral putamen.
DISCUSSION
The patient with hyperargininemia presented with certain peculiar features. These included presentation of symptoms in adult age; and late onset disease is uncommon.4 Even though patients of hyperargininemia have been reported to survive up to 37 years,3 all of these patients had had symptoms in childhood. Our patient had no symptoms in his childhood. This may be due to a partial deficiency of the arginase enzyme. Uchino et al 5 report in their genotype-phenotype correlated study that the clinical profile of the disease differs with degrees of functional activity of the protein coded by the mutant gene. This patient may have a defective but a partially functioning protein.
This patient did not have the standard arg1 gene mutations. The rs2781666 (G/T) and rs2608897 (C/T) variations noted in the patient are reported in the ITU6 population (Indian Telugu in the UK). Out of these two variations, rs2781666 was previously reported in relation with increased risk for myocardial infarction and decreased risk for pulmonary hypertension,7 where the authors have speculated that rs2781666 could result in lower arginase activity, which is in well agreement with our study of hyperargininemia as lower arginase activity would result in higher arginine levels.
Earlier reports show that crises can manifest in this disorder and precipitate all the symptoms. In this case, we premise that the antecedent binge drinking episodes by the patient, due to his new occupation as a bar-tender, may have brought out the symptoms.
This patient repeatedly picked at his clothes, tapped his bed and made groping movements with his hands. Such abnormal activities termed floccillation and has been described in delirium;8 but not in UCDs. Perhaps the elevated guanidine metabolites were responsible for this behaviour.
Aggressive type of change in personality traits have not been reported in the previous cases. Perhaps neurotoxic damage in an already developed brain presents with these symptoms.
Although epilepsy9 is one of the prominent symptoms in hyper-argininemia, our patient did not have seizures. The adult brain may be resistant to epileptogenic toxins.
In our patient, there was no nausea or vomiting, probably because of self abstinence from ammonemia generating protein-rich food.10 Most of the earlier reported patients had persistent vomiting. Adult onset encephalopathy due to arginase deficiency has not been reported widely. Chronic progressive encephalopathy may be due to unmasking of UCD, even in adults. Clinical profile of hyperargininemia due to arginase deficiency in adults may differ from its childhood presentation.
PATIENT'S CONSENT:
Written informed consent was obtained from closest relative (as patient's higher mental functions were not normal) for publication.
CONFLICT OF INTEREST:
Authors declared no conflict of interest.
AUTHORS’ CONTRIBUTION:
SN, RD, SA: Diagnosed and managed the case, drafted the manu-script, reviewed and finished the work.
REFERENCES
- Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 2012; 7:1.
- Mori A. Guanidino compounds and neurological disorders. Neurosciences 1983; 9:149-157.
- Carvalho DR, Brum JM, Speck-Martins CE, Ventura FD, Navarro MM, Coelho KE, et al. Clinical features and neurologic progression of hyperargininemia. Pediatr Neurol 2012; 46: 369-74.
- Maramattom B, Raja R, Balagopal A. Late onset arginase deficiency presenting with encephalopathy and midbrain hyperintensity. Ann Indian Acad Neurol 2016; 19:392.
- Uchino T, Snyderman SE, Lambert M, Qureshi IA, Shapira SK, Sansaricq C, et al. Molecular basis of phenotypic variation in patients with argininemia. Hum Genet 1995; 96:255-60.
- Transcript: ARG1-001 (ENST00000368087.7) - Exons - Homo sapiens - Ensembl genome browser 85 [Internet]. [cited 2016 Sep 30]. Available from: http://asia.ensembl.org/Homo_ sapiens/Transcript/Exons?db=core;g=ENSG00000118520;r=6:131573144-131584332;t=ENST00000368087.
- Trittmann JK, Nelin LD, Zmuda EJ, Gastier-Foster JM, Chen B, Backes CH, et al. Arginase I gene single-nucleotide polymorphism is associated with decreased risk of pulmonary hypertension in bronchopulmonary dysplasia. Acta Paediatr 2014; 103:e439-e43.
- Larner A. Carphologia, or floccillation. Curr Treat Options Neurol 2007; 7:25.
- Schlune A, Vom Dahl S, Häussinger D, Ensenauer R, Mayatepek E. Hyperargininemia due to arginase I deficiency: The original patients and their natural history, and a review of the literature. Amino Acids 2015; 47:1751-62.
- Prasad AN, Breen JC, Ampola MG, Rosman NP. Argininemia: A treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. J Child Neurol 1997; 12:301-9.