A Case with Neonatal-onset Type 2 Neuronal Ceroid Lipofuscinosis: A Novel Mutation
By Ozgun Uygur1, Mehmet Yekta Oncel1,2, Pinar Gencpinar3, Merve Saka Guvenc4, Nihal Olgac Dundar3Affiliations
doi: 10.29271/jcpsp.2020.05.543ABSTRACT
Neuronal ceroid lipofuscinosis (NCL) is a lysosomal storage disorder that causes progressive neurodegenerative disease as a result of storage in neurons and other cells. Late infantile type (NCL Type 2) of NCL, which is the most common neurodegenerative disease in childhood, is characterised by a homozygous mutation in the tripeptidyl peptidase-1 (TPP-1) gene. A male infant was referred to our neonatal intensive care unit (NICU) on 26th day of life with a diagnosis of metabolic disease. He was intubated. He was hypotonic and newborn reflexes were not present. Cranial magnetic resonance (MR) imaging revealed severe atrophy and delayed myelination of cerebellum and cerebral hemispheres. A novel homozygous pathological mutation was detected in exon 9 of the TPP-1 gene. With this case, it should be kept in mind that NCL may rarely start early in neonatal period and should be suspected in newborns with cerebral and cerebellar atrophy for early diagnosis.
Key Words: Hypotonia, Lysosomal storage diseases, Metabolic disease, Neuronal ceroid lipofuscinosis, Newborn.
INTRODUCTION
Neuronal ceroid lipofuscinosis (NCL) is a lysosomal storage disorder that causes progressive neurodegenerative disease as a result of storage in neurons and other cells. Late infantile type (NCL Type 2) of NCL, which is the most common neurodegenerative disease in childhood, is characterised by a homozygous mutation in the TPP-1 gene.1 First symptoms typically occur between 2-4 years of age. After the onset of seizures, the acquired motor/language and cognitive skills are gradually lost. The patients usually become severely disabled and bedridden at the age of 6 years, requiring nursing care. Here we report a case of neonatal-onset NCL type 2 diagnosed for the first time with a novel mutation.
CASE REPORT
A male infant was delivered by a 39-year mother by cesarean section at 38 weeks of gestation with a birth weight of 2.605 Kg. The baby was referred from an external centre to our neonatal intensive care unit (NICU) on his 26th day of life with a diagnosis of metabolic disease.
No consanguinity was present between the parents; and the family also had a healthy girl. His mother reported decreased baby movements during her pregnancy. On physical examination, his body weight was 2.330 Kg (3rd percentile), length was 42 centimeters (3rd percentile), and head circumference was 29 centimeters (<3rd percentile). He was intubated. He was hypotonic and newborn reflexes were not present. Cranial magnetic resonance (MR) imaging revealed severe atrophy and delayed myelination of cerebellum and cerebral hemispheres (Figures 1a, 1b). Electroneuromyography revealed no myopathic discharge and no primary muscle fiber involvement. Muscle biopsy was normal. No gene mutation was detected for spinal muscular atrophy. A homozygous pathological mutation of c.1145G>A (p.Ser382Asn) (rs761448855) was detected in exon 9 of the TPP-1 gene. The mother, father and his sister were heterozygous for the same mutation (Figure 2). The patient was diagnosed as NCL type 2 with his clinical findings and mutation. The patient died on the 132th day of his life in NICU. Genetic counselling was recommended to the family for subsequent pregnancies.
DISCUSSION
NCL is the most common neurodegenerative lysosomal disorder group in children. NCLs are classified as infantile NCL, late infantile NCL (LINCL), juvenile NCL, adult NCL, and other variant forms.2,3
NCL2 gene, which encodes the tripeptidyl-peptidase 1, is located on chromosome 11p15; and mutation in this gene may lead to TPP-1 deficiency, resulting in symptoms of classical LINCL.3
NCL is a neurodegenerative disease with progressive neuronal loss in the cerebral-cerebellar cortex and retina. Up to now, seven sub-types of NCL have been identified. LINCL is the most heterogeneous type and clinical findings of this type start between 2.5-5 years of age. Loss of acquired abilities, ataxia, epilepsy, and vision loss are the most important findings.4 While in most cases, epilepsy is the first symptom, mental deterioration, ataxia and myoclonic jerks start usually after a couple of months; and then visual failure appears after the age of four.5 In our case, interestingly, clinical findings were observed in the neonatal period with hypotonia, absence of newborn reflexes and the patient was lost at his 5th month of life.
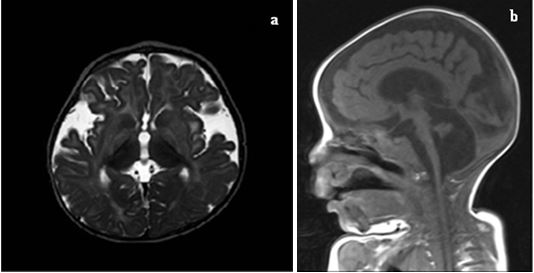
Figure 1: (a,b) Cranial MR images of the patient showing severe cellebellar and cerebral atrophy.
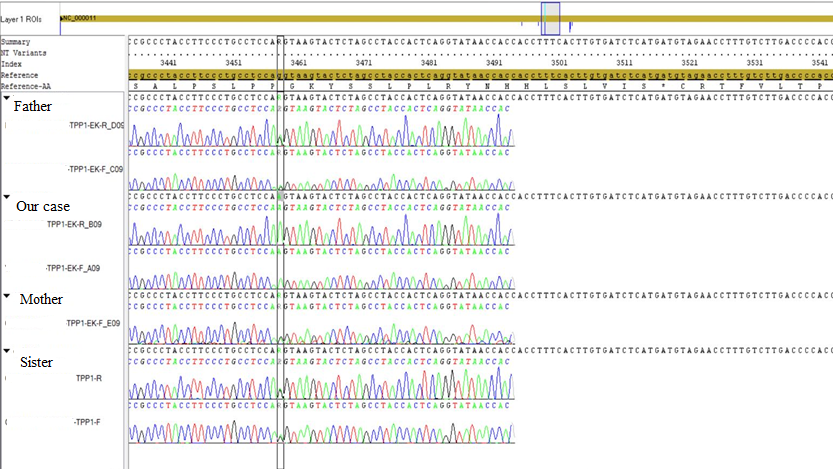 Figure 2. Genetic results of the family.
Figure 2. Genetic results of the family.
In classic LINCL, the seizures may be different from each other such as partial, generalised tonic-clonic, secondary generalised or absences. At the beginning of the disease, mild cerebral atrophy may be recorded; but after that, progressive brain atrophy is noticed.6 In our case, as expected, severe atrophy and delayed myelination of cerebellum and cerebral hemispheres were detected in cranial MR imaging.
When clinical and laboratory findings suggest NCL, the TPP1 enzyme activity should be analysed.7 Up-to-date, 54 mutations and 22 polymorphisms in NCL 2 gene have been recorded.8 Interestingly, in our case, the diagnosis of NCL type 2 was made with homozygous pathological mutation in the TPP-1 gene with a mutation of c.1145G> A (p.Ser382Asn), which is expected in late-onset NCL. A heterozygous pattern of the same mutation was detected in the mother, father and his healthy sister. To our knowledge, this mutation is novel and yet not reported in literature; but this mutation is called as ‘likely pathogenic’ in the Varsome database. This naming is done because it meets the criteria of PM1, PM2, PM5, PP2, and PP3 in the ACMG classification.
With this case, it should be kept in mind that NCL may rarely start early in newborns; and in the presence of cerebral and cerebellar atrophy, NCL should be considered for early diagnosis and maybe for total curative treatment in the future.
PATIENT'S CONSENT:
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
CONFLICT OF INTEREST:
Authors declared no conflict of interest.
AUTHORS’ CONTRIBUTION:
OU: Drafted the initial manuscript.
OU, MYO: Reviewed and revised the manuscript.
PG, MSG, NOD: Designed the data collection instruments, collected data and carried out the initial analyses.
OU, MYO: Conceptualised and designed the study, coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content.
All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
REFERENCES
- Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases: Clinical perspectives. Biochim Biophys Acta 2013; 1832:1801-6.
- Mole SE, Williams RE, Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005; 6:107-26.
- Yu F, Liu XM, Chen YH, Zhang SQ, Wang K. A novel CLN2/TPP1 mutation in a patient with late infantile neuronal ceroid lipofuscinosis. Neurol Sci 2015; 36:1917-9.
- Yiş U, Kurul SH, Özoğul C, Dirik E. Late infantile neuronal ceroid lipofuscinosis: A case report. Turk Arch Ped 2010; 45:155-7.
- Santavuori P, Lauronen L, Kirveskari E, Aberg L, Sainio K, Autti T. Neuronal ceroid lipofuscinoses in childhood. Neurol Sci 2000; 21:S35-S41.
- Dunn D. CT in ceroid lipofuscinosis. Neurology 2000; 37: 1025-6.
- Fietz M, AlSayed M, Burke D, Cohen-Pfeffer J, Cooper JD, Dvořáková L, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab 2016; 119:160-7.
- Bessa C, Teixeira CA, Dias A, Alves M, Rocha S, Lacerda L, et al. CLN2/TPP1 deficiency: The novel mutation IVS7-10A[G causes intron retention and is associated with a mild disease phenotype. Mol Genet Metab 2008; 93:66-73.